Pharmacogenetics of Vascular Risk Factors in Alzheimer’s Disease

 ,
,  ,
, 
Abstract
1. Introduction
2. Genetic Determinants of the Pharmacogenomic Network
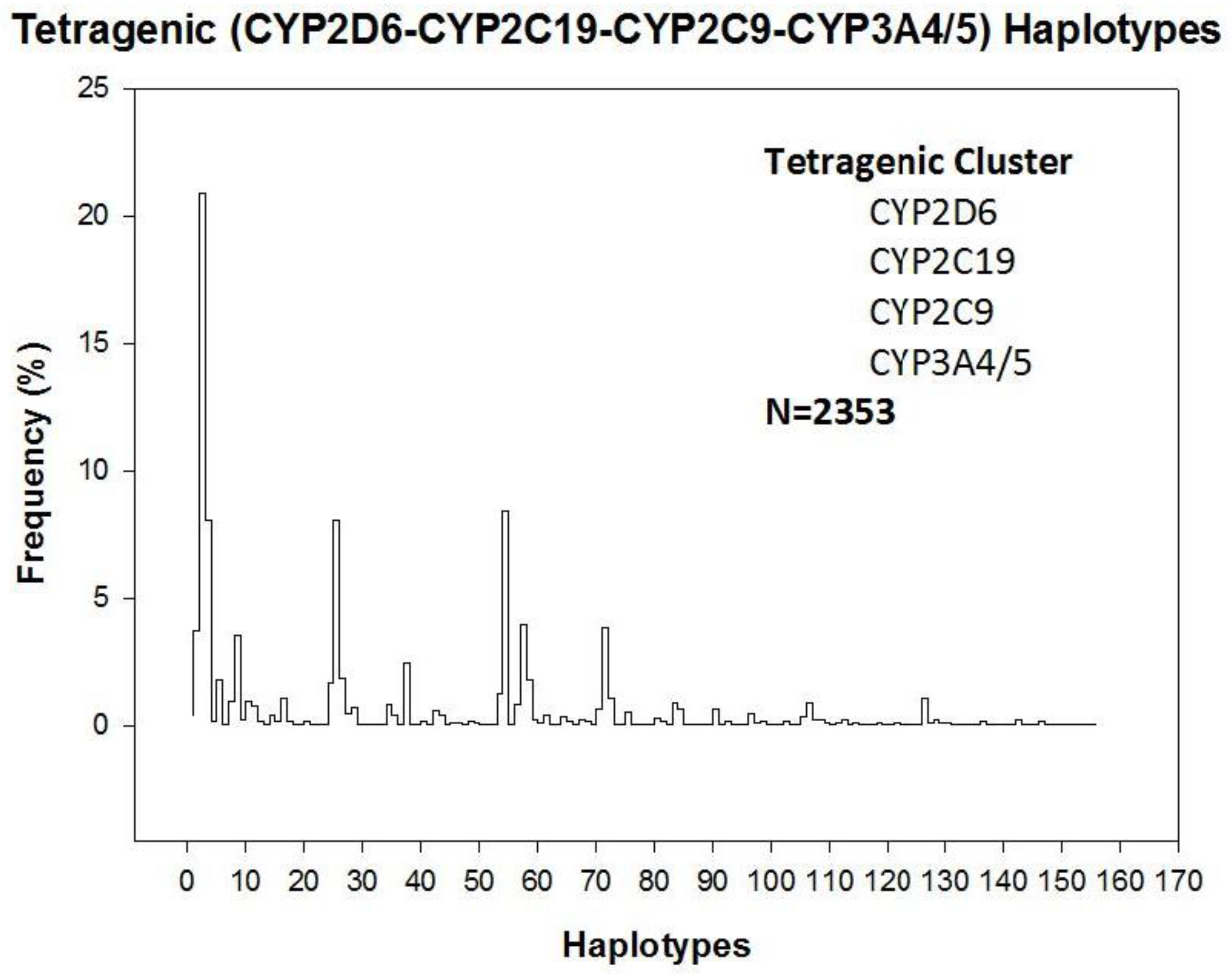
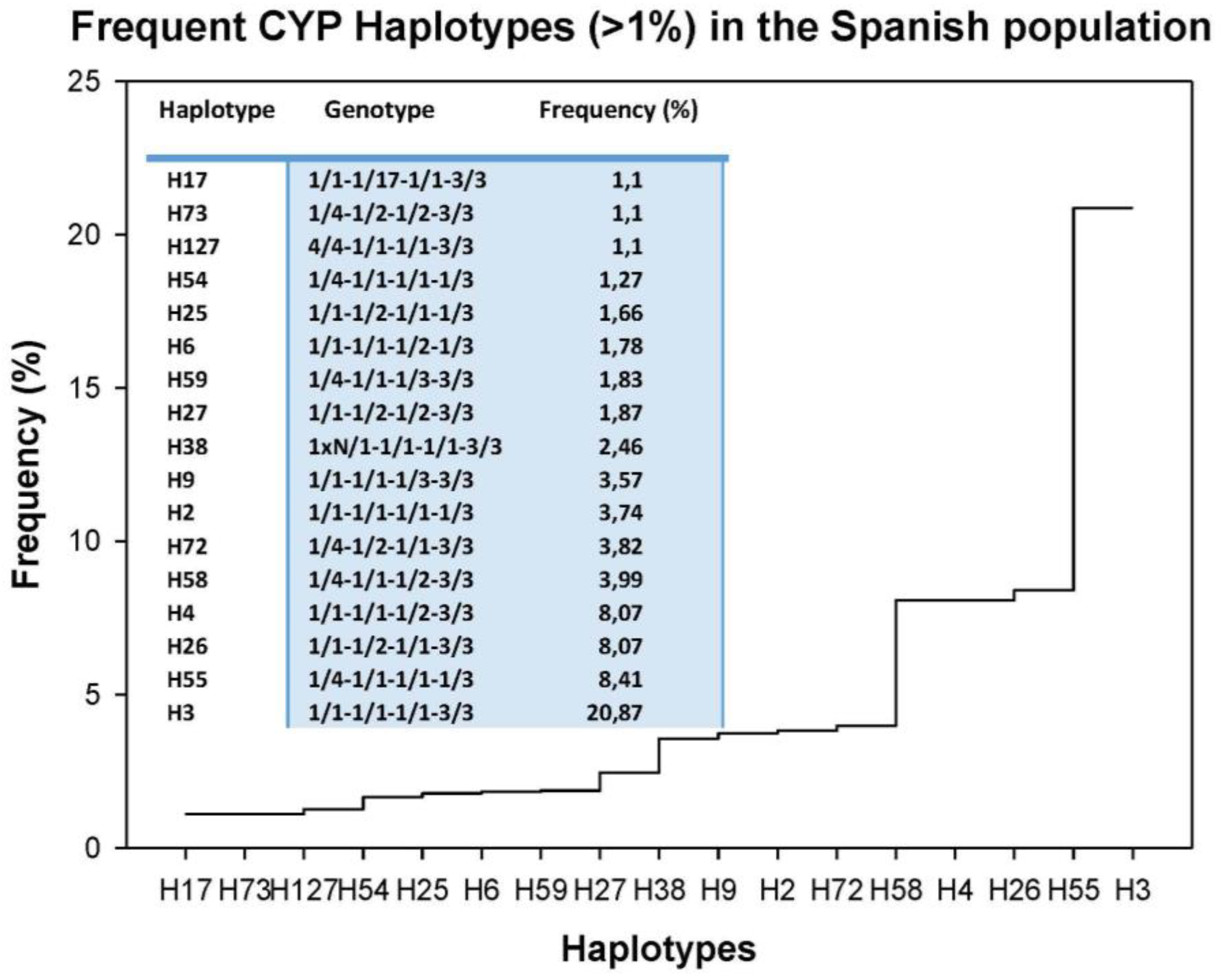
3. Drug Metabolism
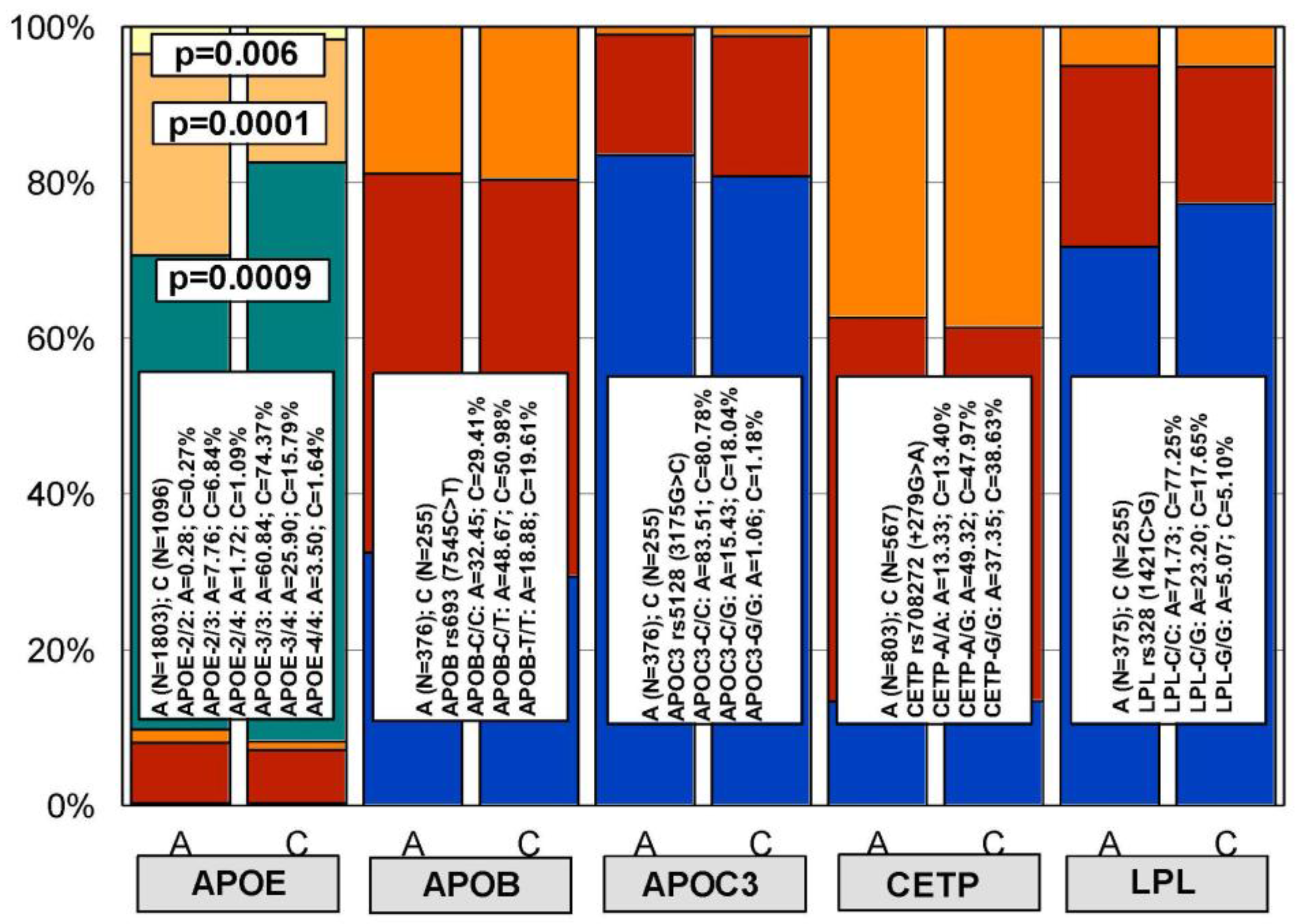
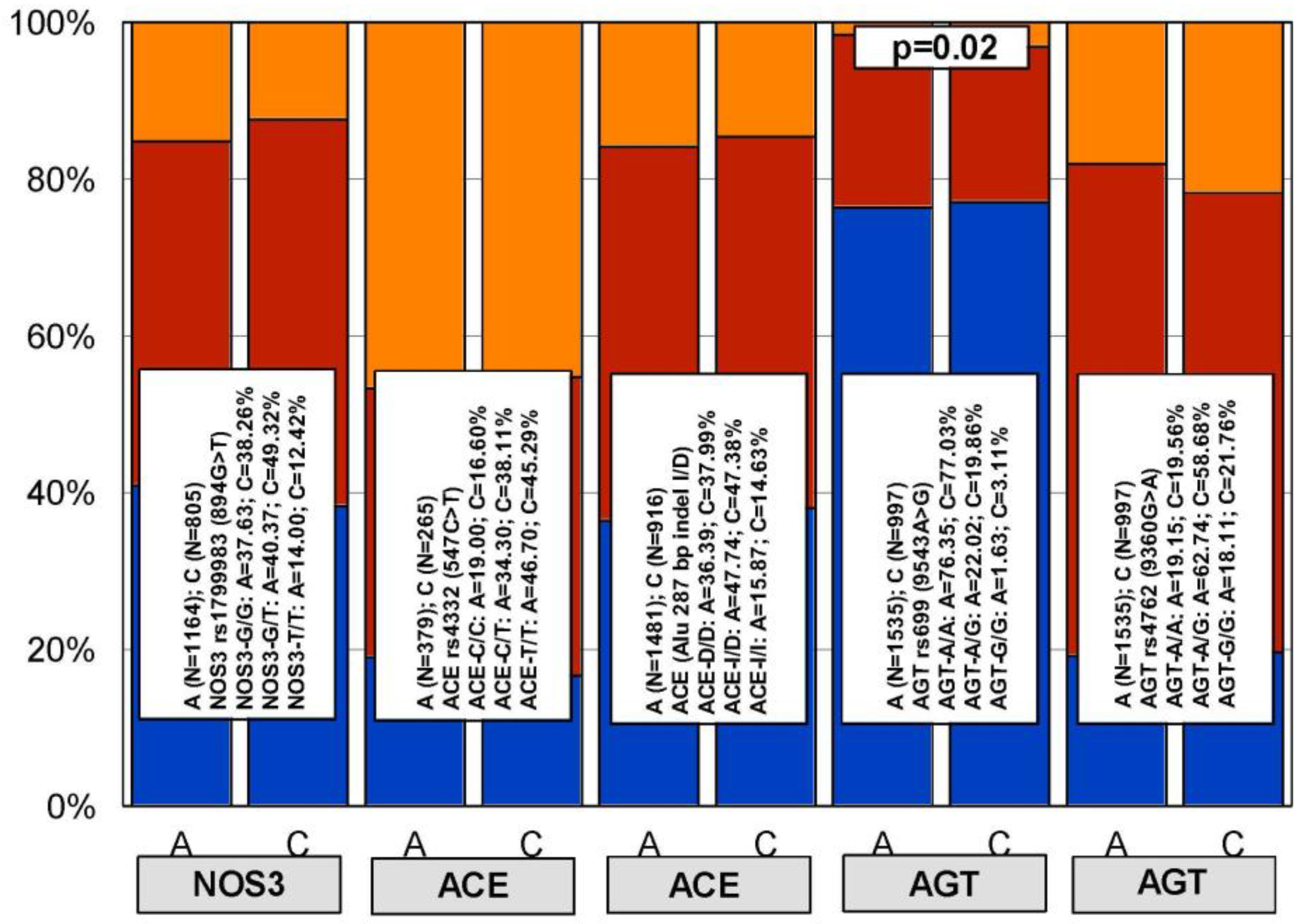
4. Genetic Determinants of Lipid Metabolism and Vascular Function
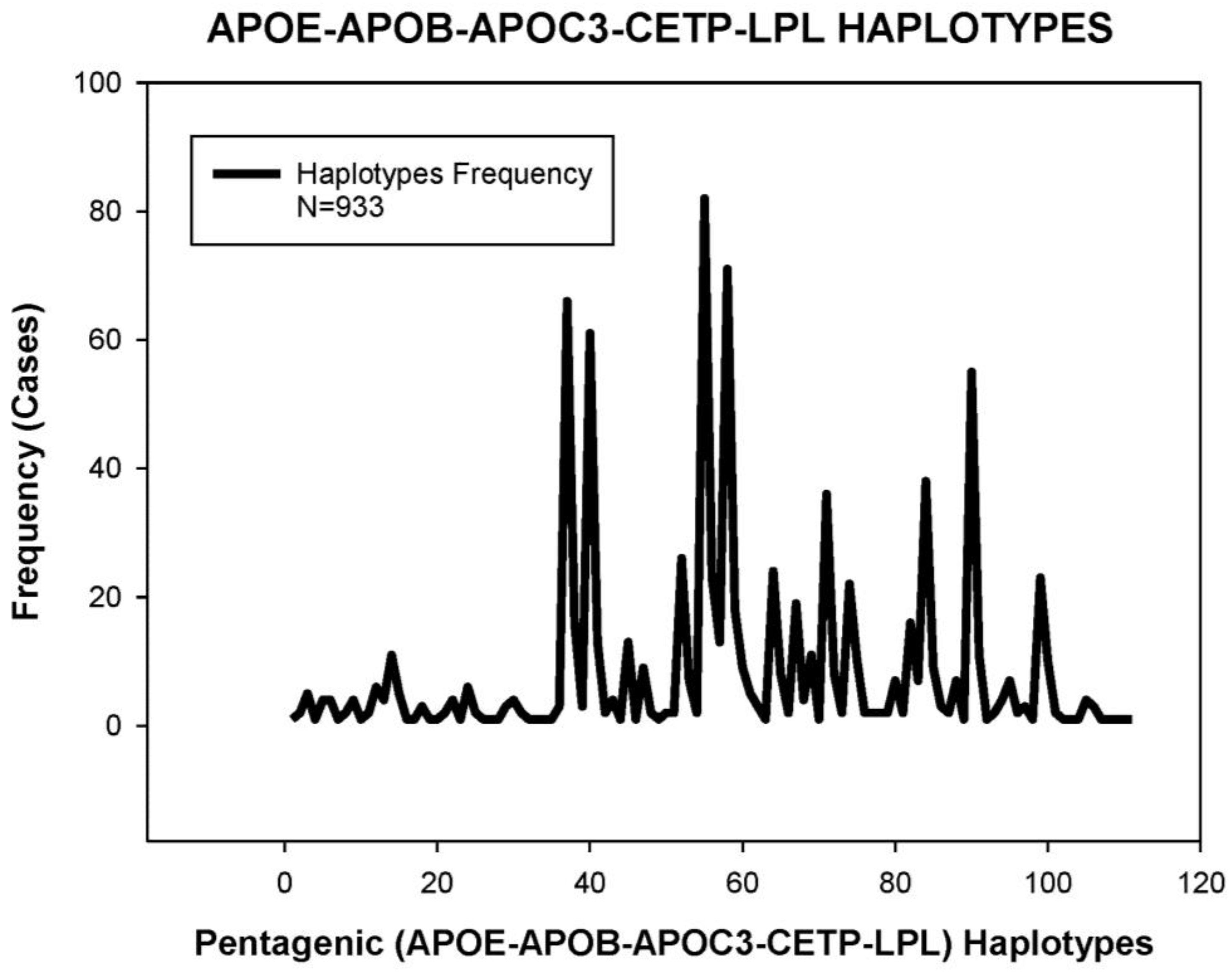
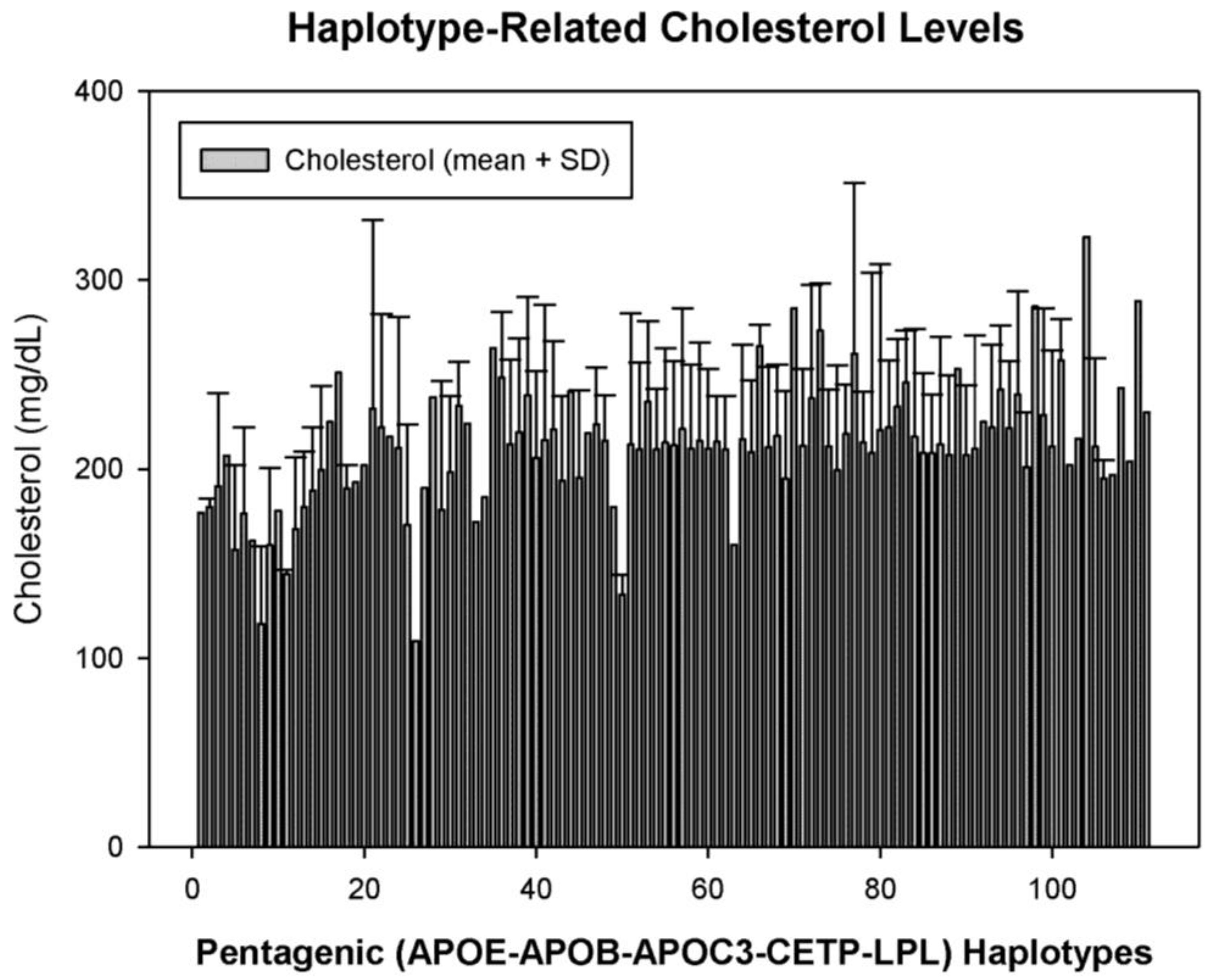
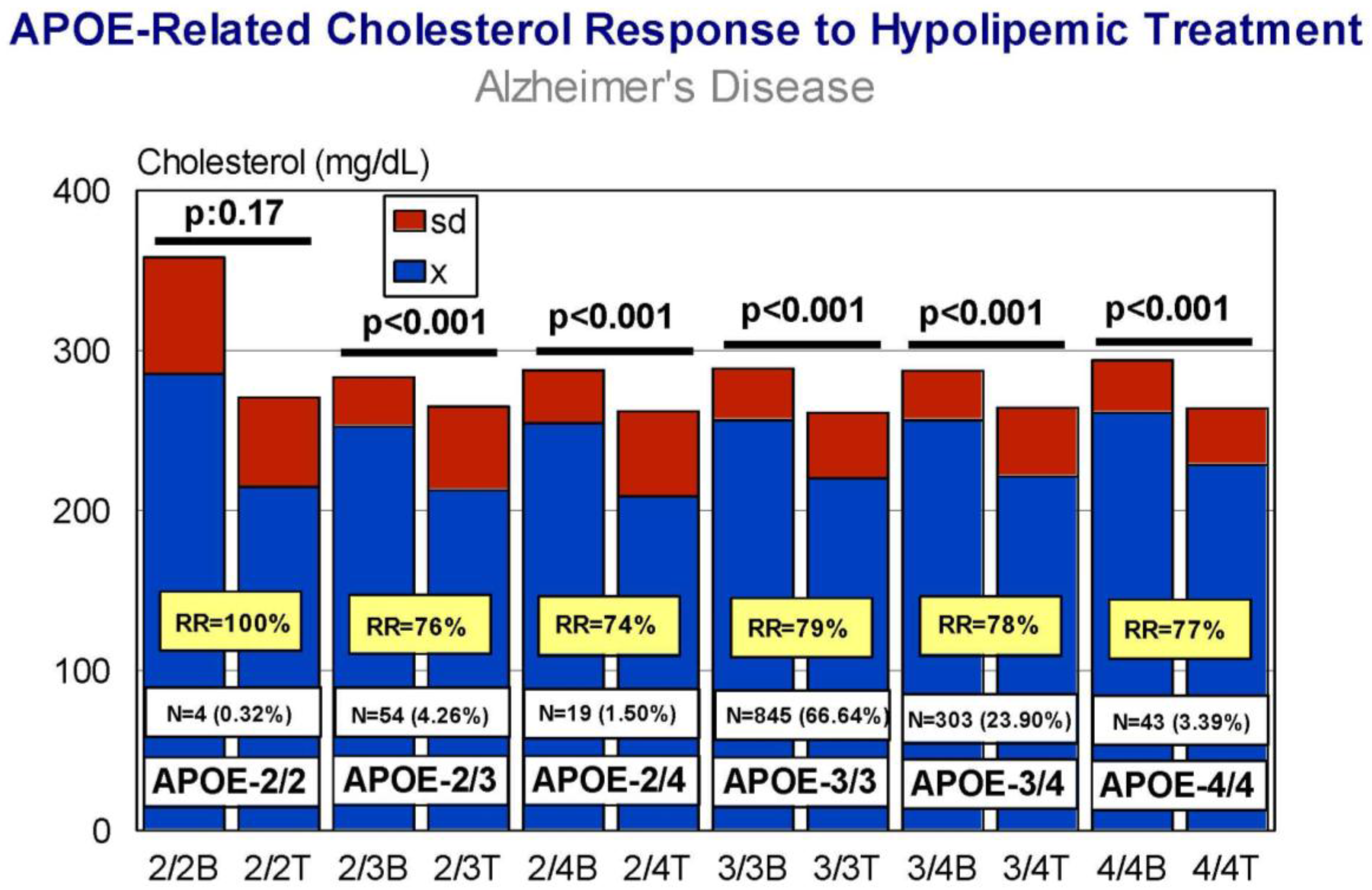
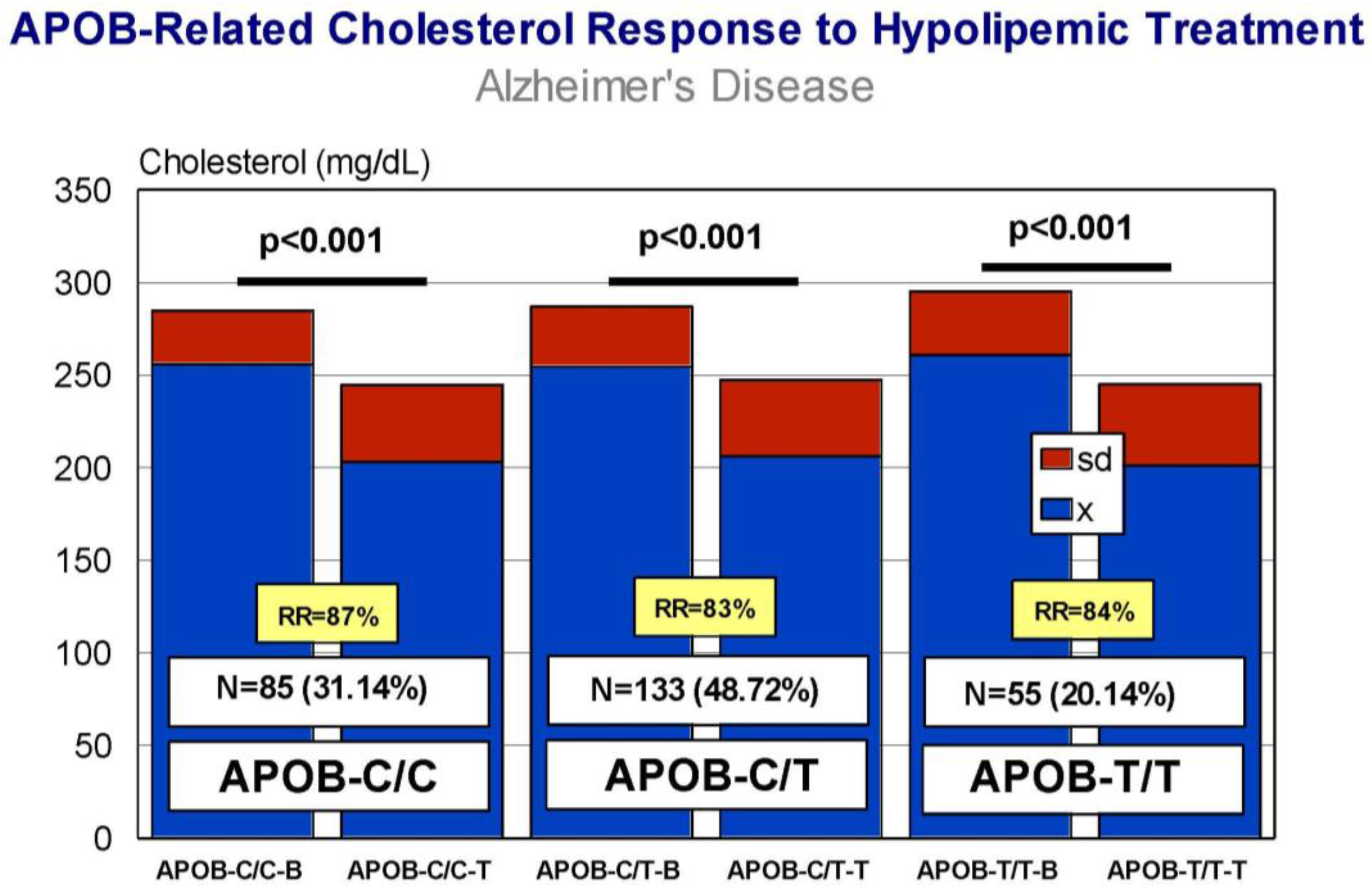
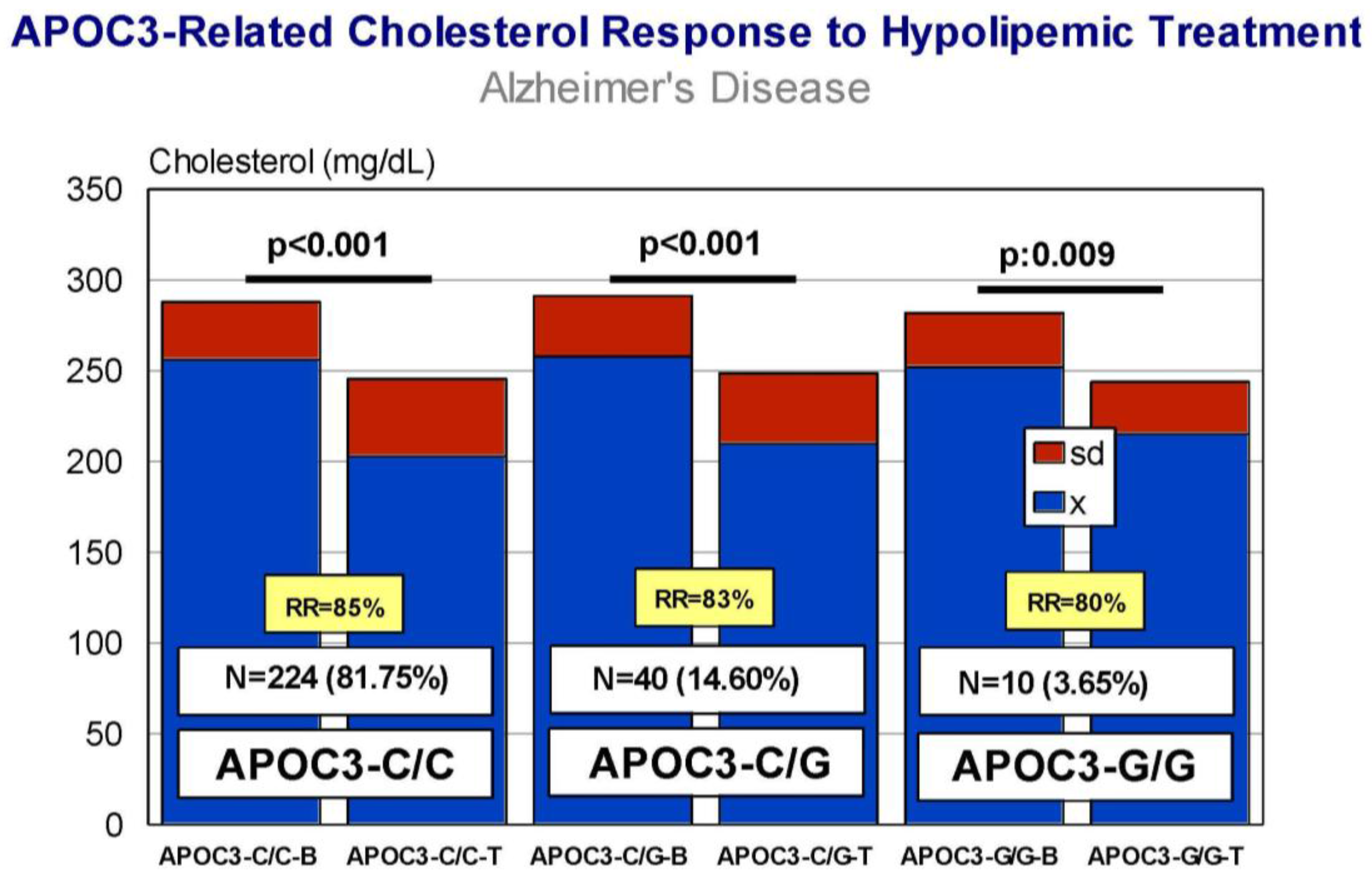
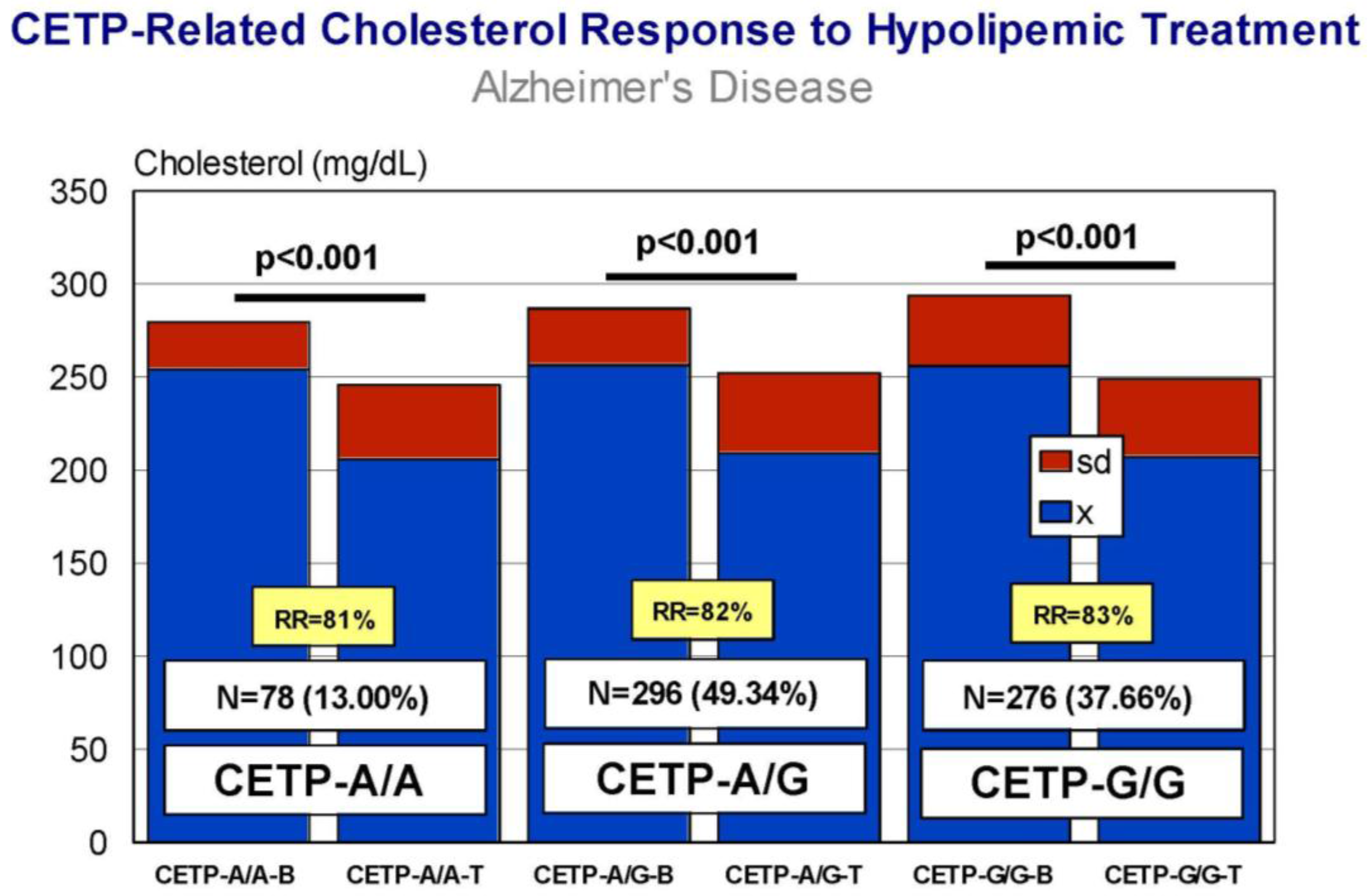
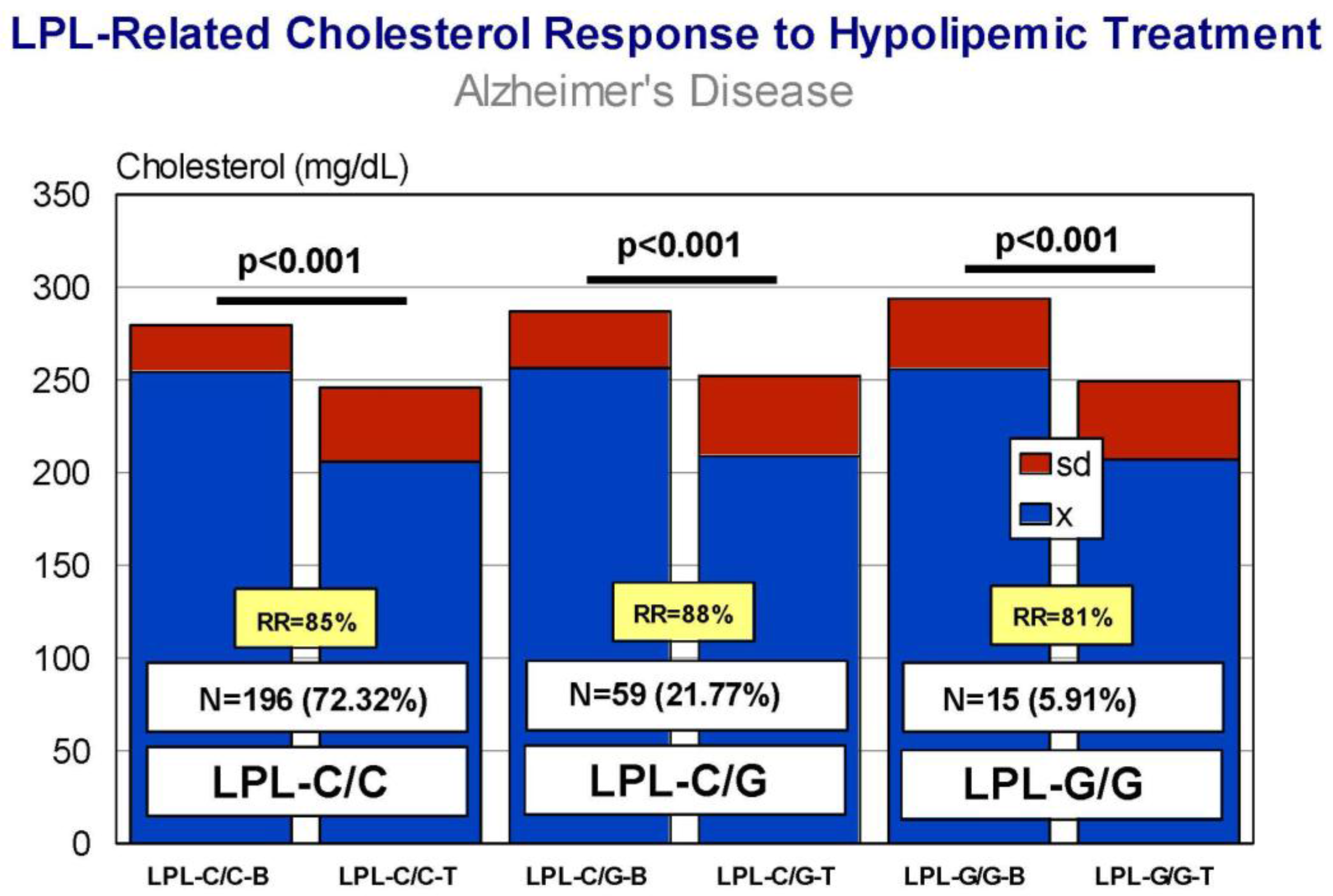
5. Pharmacogenetics of Hypercholesterolemia in Alzheimer’s Disease
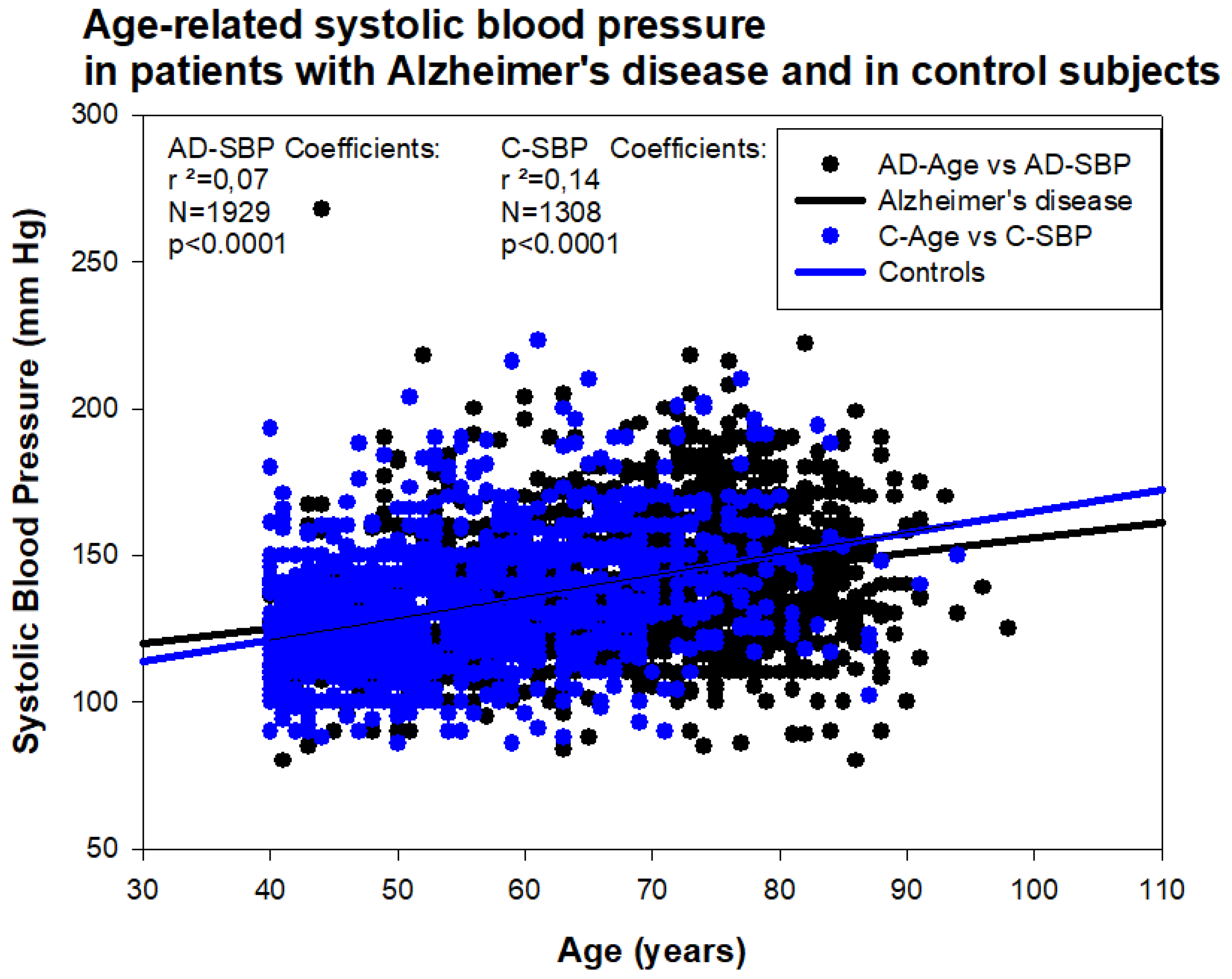
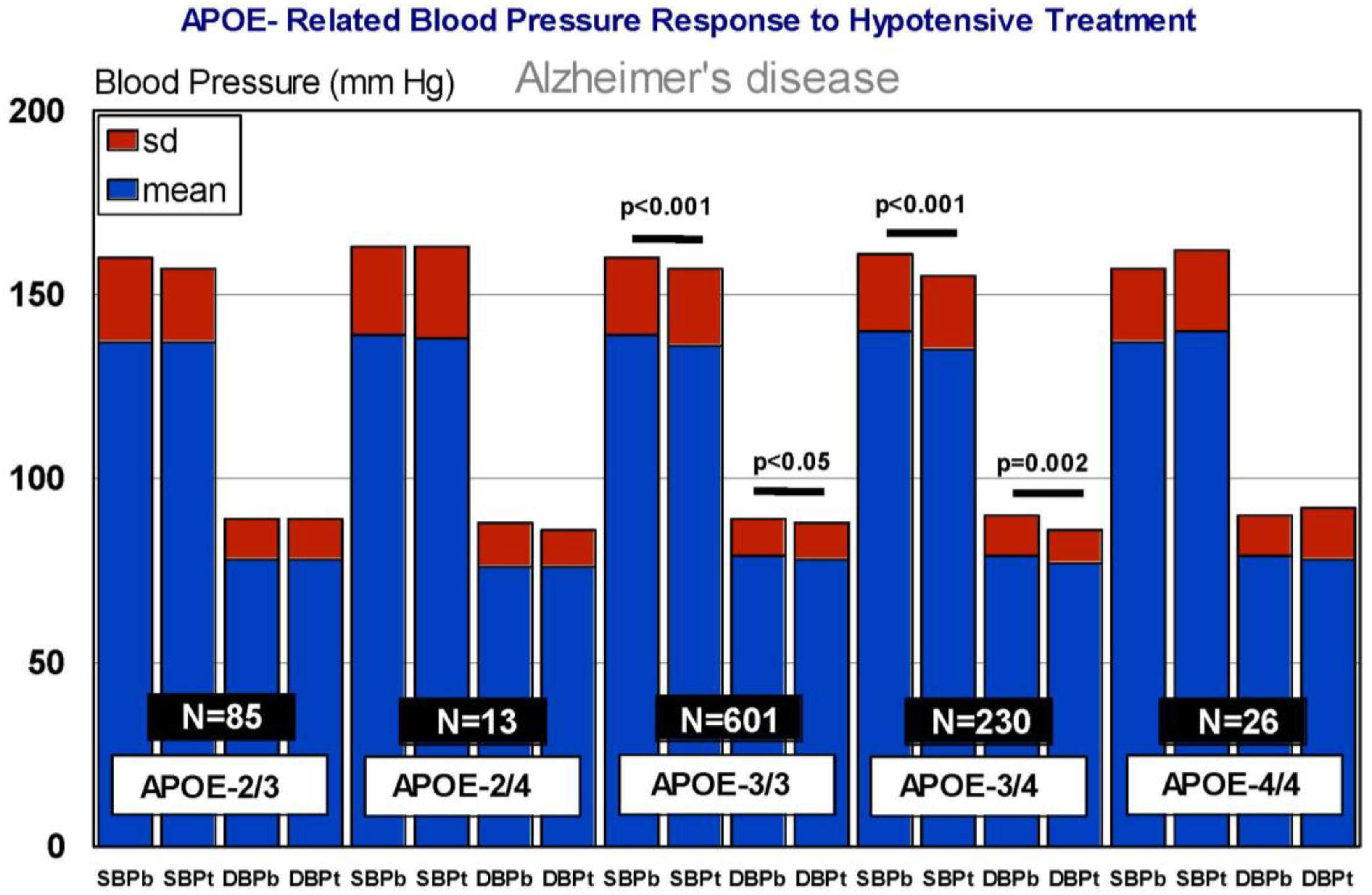
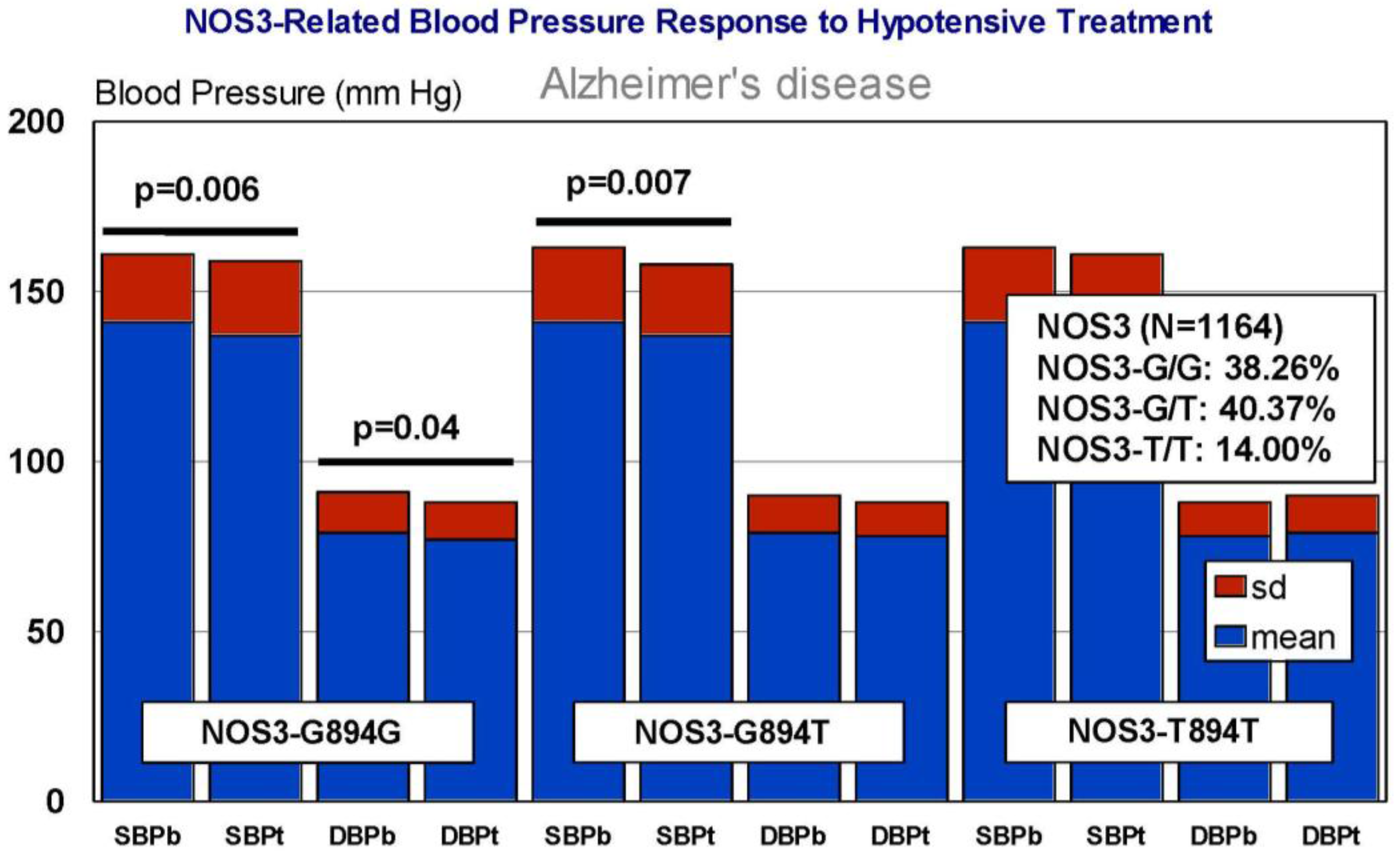
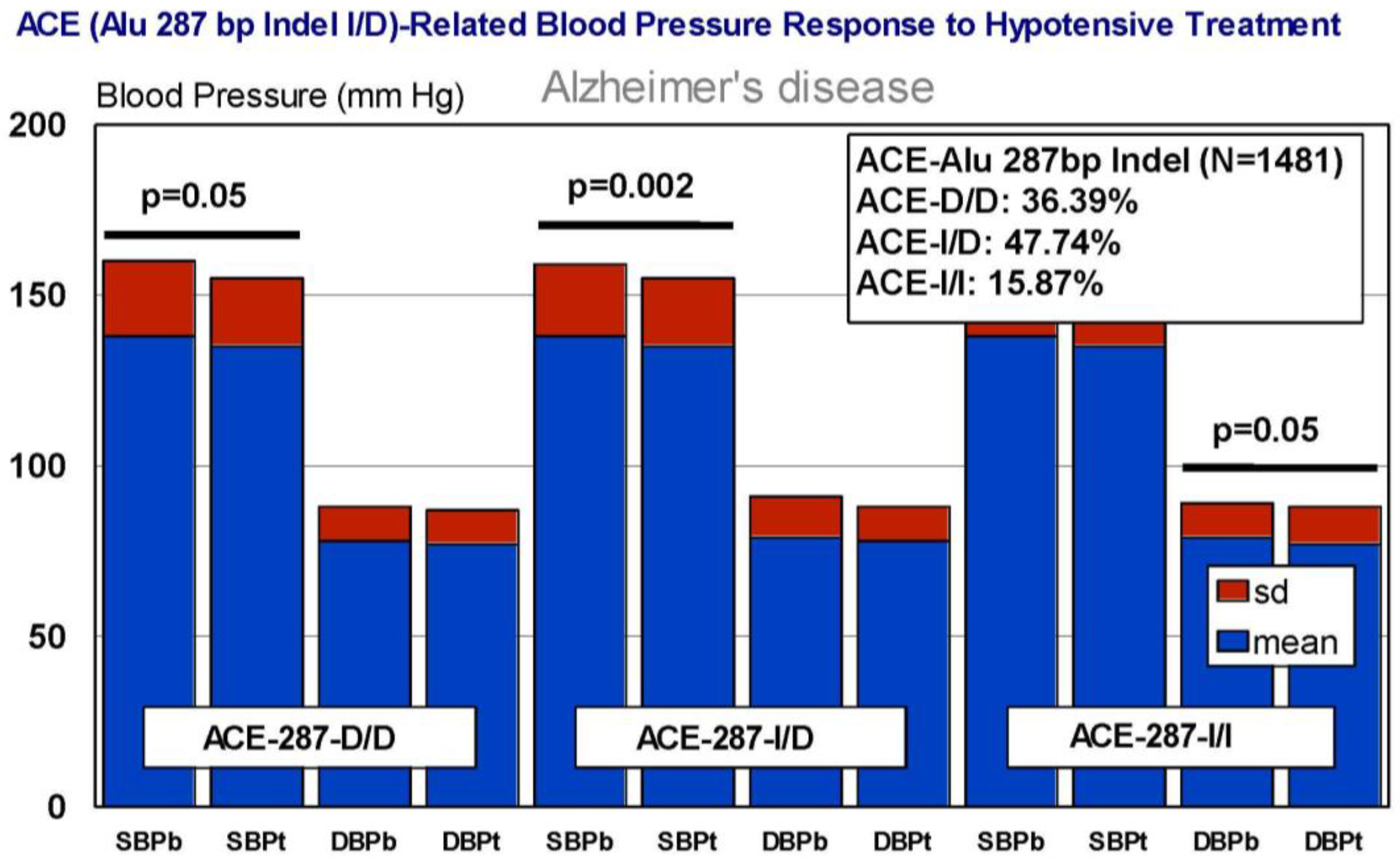
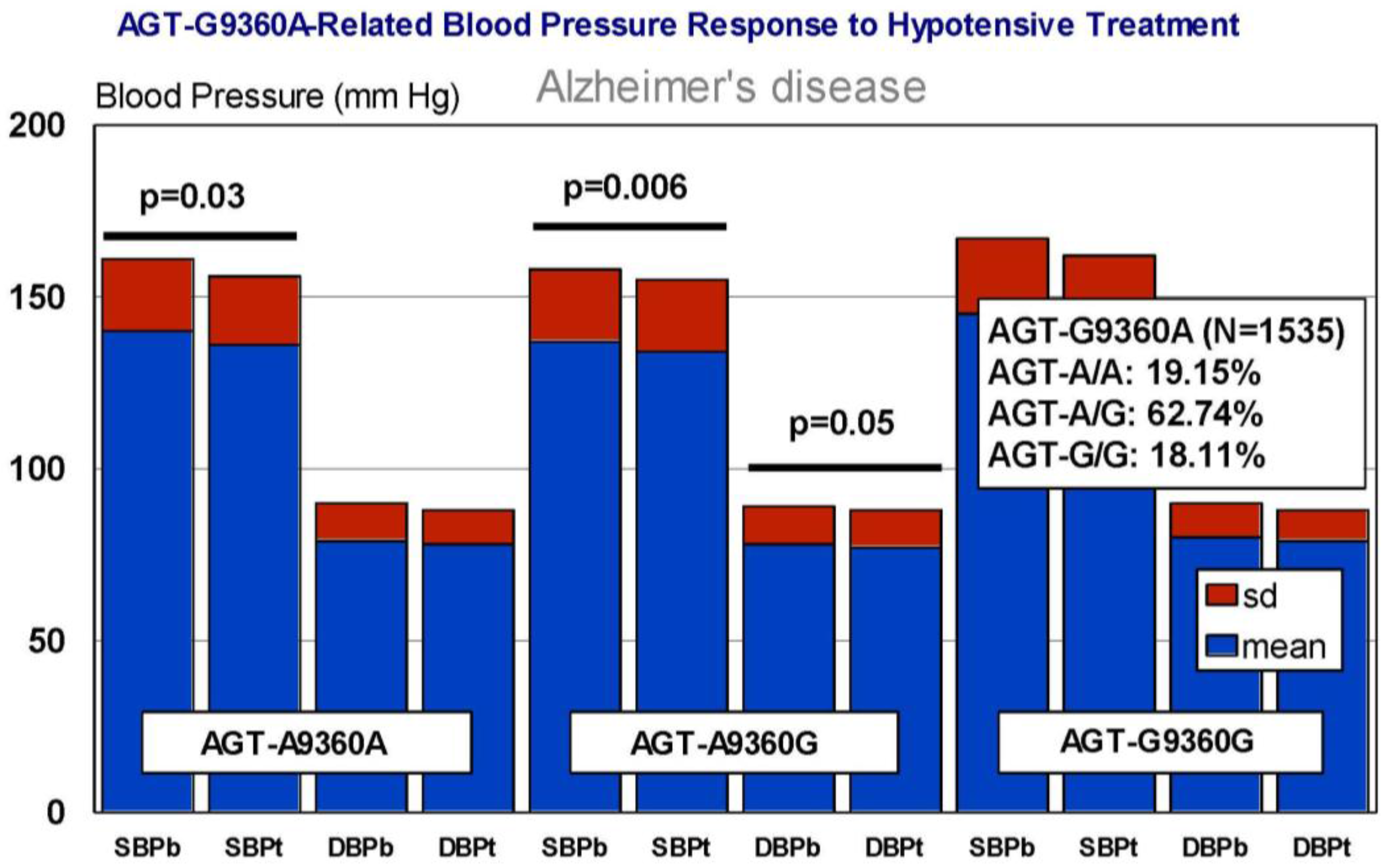
6. Pharmacogenetics of Hypertension in Alzheimer’s Disease
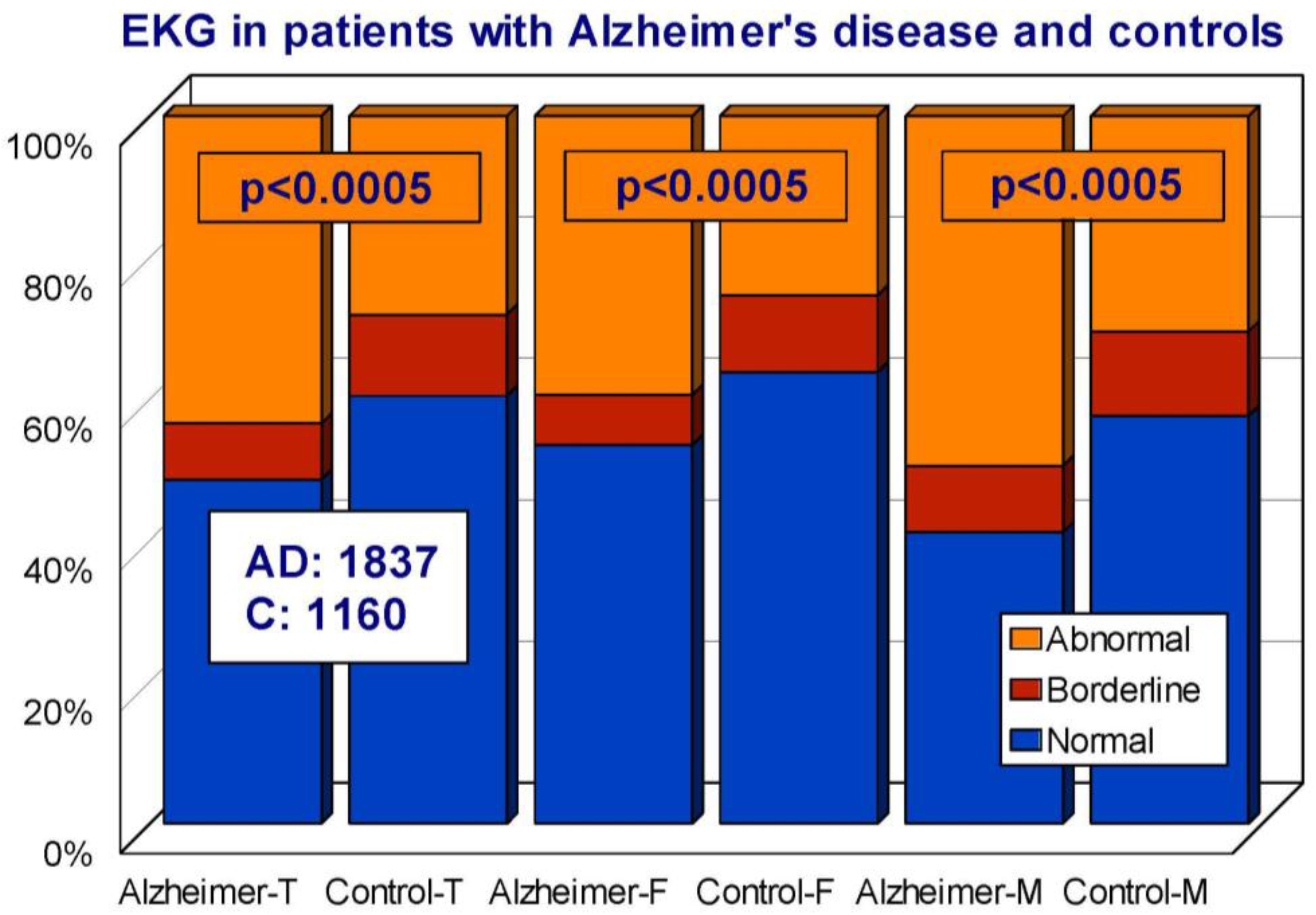
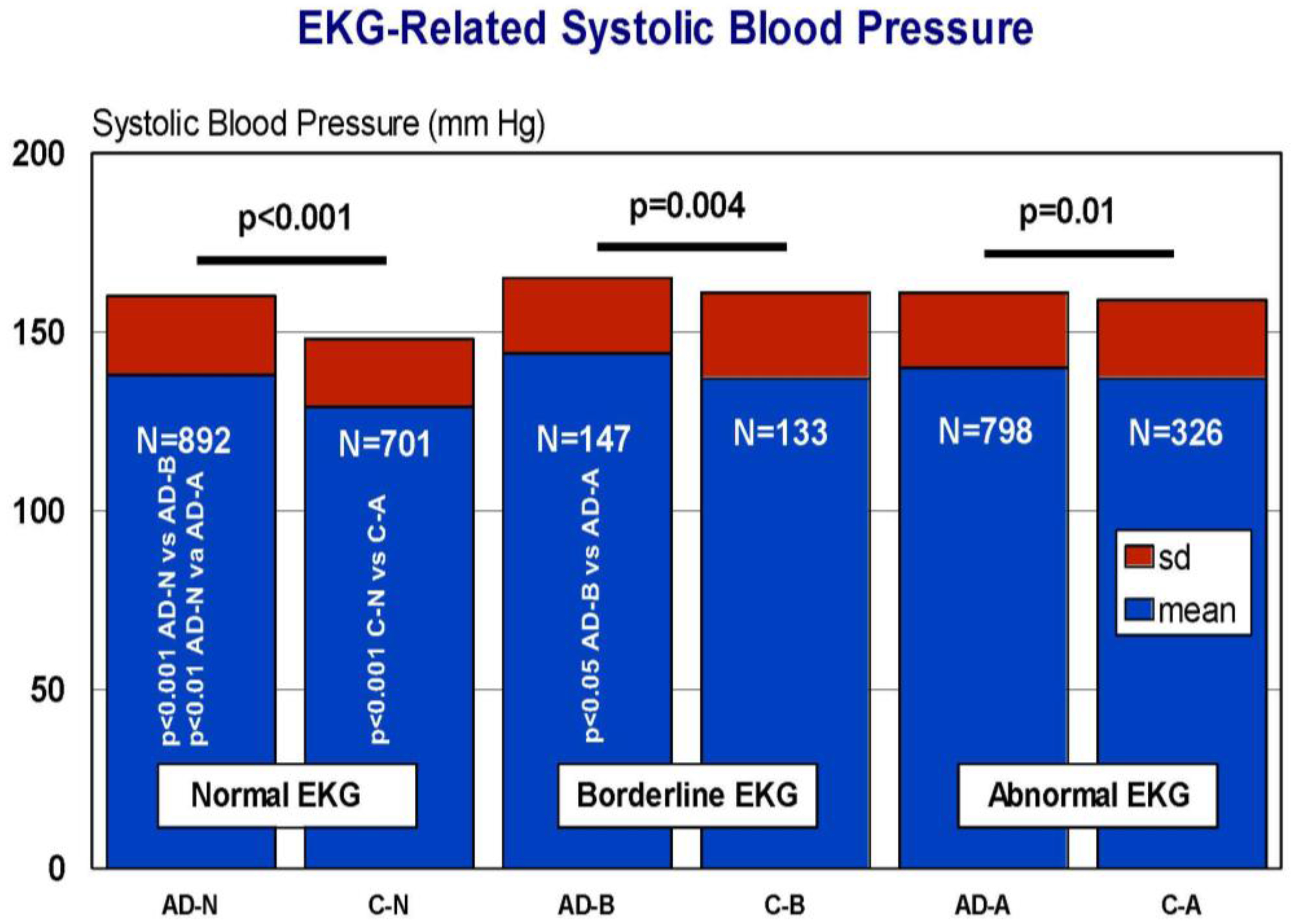
7. EKG Conditions and Blood Pressure in Alzheimer’s Disease
8. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Cacabelos, R. The application of functional genomics to Alzheimer’s disease. Pharmacogenomics 2003, 4, 597–621. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Molecular pathology and pharmacogenomics in Alzheimer’s disease: Polygenic-related effects of multifactorial treatments on cognition, anxiety, and depression. Methods Find. Exp. Clin. Pharmacol. 2007, 29 (Suppl. A), 1–91. [Google Scholar] [PubMed]
- Cacabelos, R. Pharmacogenomics and therapeutic strategies for dementia. Expert Rev. Mol. Diagn. 2009, 9, 567–611. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Corzo, L.; Amado, L.; Pichel, V.; Lombardi, V.; Kubota, Y. Phenotypic profiles and functional genomics in Alzheimer’s disease and in dementia with a vascular component. Neurol. Res. 2004, 26, 459–480. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Cerebral Perfusion Enhancing Interventions: A New Strategy for the Prevention of Alzheimer Dementia. Brain Pathol. 2016, 26, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Goldgaber, D.; Roses, A.D.; Vostrov, A.; Matsuki, H.; Torrellas, C.; Cacabelos, P.; Corzo, D.; Carril, J.C.; Fernández-Novoa, L.; et al. Gene interactions in the Pharmacogenomics of Alzheimer’s Disease. Sciforschen Genet. Gene Ther. 2015, 1. [Google Scholar] [CrossRef]
- Cacabelos, R.; Goldgaber, D.; Vostrov, A.; Matsuki, H.; Torrellas, C.; Corzo, D.; Carril, J.C.; Roses, A.D. APOE-TOMM40 in the Pharmacogenomics of dementia. J. Pharmacogenom. Pharmacoproteom. 2014, 5, 135. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C.; Carrera, I. Opportunities in Pharmacogenomics for the treatment of Alzheimer’s Disease. Future Neurol. 2015, 10, 229–252. [Google Scholar] [CrossRef]
- Taipale, H.; Koponen, M.; Tanskanen, A.; Tolppanen, A.M.; Tiihonen, J.; Hartikainen, S. Drug use in persons with and without Alzheimer’s disease aged 90 years or more. Age Ageing 2016, 45, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Cacabelos, P.; Torrellas, C.; Tellado, I.; Carril, J.C. Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Methods Mol. Biol. 2014, 1175, 323–556. [Google Scholar] [PubMed]
- Cacabelos, R.; Torrellas, C.; Carrera, I.; Cacabelos, P.; Corzo, L.; Fernández-Novoa, L.; Tellado, I.; Carril, J.C.; Aliev, G. Novel Therapeutic Strategies for Dementia. CNS Neurol. Disord. Drug Targets 2016, 15, 141–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, Z.; Lin, E.; Zhao, W.; Qian, X.; Freire, D.; Bilski, A.E.; Cheng, A.; Vempati, P.; Ho, L.; et al. Unintended effects of cardiovascular drugs on the pathogenesis of Alzheimer’s disease. PLoS ONE 2013, 8, e65232. [Google Scholar] [CrossRef] [PubMed]
- Montastruc, F.; Gardette, V.; Cantet, C.; Piau, A.; Lapeyre-Mestre, M.; Vellas, B. Potentially inappropriate medication use among patients with Alzheimer disease in the REAL.FR cohort: Be aware of atropinic and benzodiazepine drugs! Eur. J. Clin. Pharmacol. 2013, 69, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wen, G.; Li, Y.; Liu, C. The occurrence of cerebrovascular atherosclerosis in Alzheimer’s disease patients. Clin. Interv. Aging 2013, 8, 581–584. [Google Scholar] [PubMed]
- Chen, Y.; Sillaire, A.R.; Dallongeville, J.; Skrobala, E.; Wallon, D.; Dubois, B.; Hannequin, D.; Pasquierthe, F. Lille YOD Study Group. Low Prevalence and Clinical Effect of Vascular Risk Factors in Early-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C.; Teijido, O.; Carril, J.C. Pharmacogenetic considerations in the treatment of Alzheimer’s Disease. Pharmacogenomics 2016, 17, 1041–1074. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Carril, J.C.; Cacabelos, P.; Teijido, O.; Goldgaber, D. Pharmacogenomics of Alzheimer’s Disease: Genetic determinants of phenotypic variation and therapeutic outcome. J. Genom. Med. Pharmacogenom. 2016, 1, 151–209. [Google Scholar]
- Cacabelos, R.; Fernández-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and aging. Methods Find. Exp. Clin. Pharmacol. 2005, 27 (Suppl. A), 1–573. [Google Scholar] [PubMed]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Cruchaga, C.; Goate, A. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Etcheverría, I.; Fernández-Novoa, L.; Lombardi, V.; Cacabelos, R.; Vigo, C. Vaccine Development to Treat Alzheimer’s Disease Neuropathology in APP/PS1 Transgenic Mice. Int. J. Alzheimers Dis. 2012, 376138. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Etcheverría, I.; Fernández-Novoa, L.; Lombardi, V.R.M.; Lakshmana, M.K.; Cacabelos, R.; Vigo, C. A comparative evaluation of a novel vaccine in APP/PS1 mouse models of Alzheimer’s Disease. BioMed Res. Int. 2015, 807146. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Martínez-Bouza, R.; McKay, A.; Carril, J.C.; Lombardi, V.; Corzo, L.; Carrera, I.; Tellado, I.; Nebril, L.; et al. Future trends in the pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6 variants. Pharmaceuticals 2010, 3, 3040–3100. [Google Scholar] [CrossRef]
- Cacabelos, R.; Llovo, R.; Fraile, C.; Fernández-Novoa, L. Pharmacogenetic aspects of therapy with cholinesterase inhibitors: The role of CYP2D6 in Alzheimer’s disease pharmacogenetics. Curr. Alzheimer Res. 2007, 4, 479–500. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C. Epigenetics of aging and Alzheimer’s disease: Implications for pharmacogenomics and drug response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef] [PubMed]
- Clarelli, F.; Mascia, E.; Santangelo, R.; Mazzeo, S.; Giacalone, G.; Galimberti, D.; Fusco, F.; Zuffi, M.; Fenoglio, C.; Franceschi, M.; et al. CHRNA7 Gene and Response to Cholinesterase Inhibitors in an Italian Cohort of Alzheimer’s Disease Patients. J. Alzheimers Dis. 2016, 52, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Myung, W.; Lim, S.W.; Kang, H.S.; Kim, S.; Won, H.H.; Carroll, B.J.; Kimet, D.K. Association of the choline acetyltransferase gene with responsiveness to acetylcholinesterase inhibitors in Alzheimer’s disease. Pharmacopsychiatry 2015, 48, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Braga, I.L.; Silva, P.N.; Furuya, T.K.; Santos, L.C.; Pires, B.C.; Mazzotti, D.R.; Bertolucci, P.H.; Cendoroglo, M.S.; Smith, M.C. Effect of APOE and CHRNA7 genotypes on the cognitive response to cholinesterase inhibitor treatment at different stages of Alzheimer’s disease. Am. J. Alzheimers Dis. Other Dement. 2015, 30, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Sokolow, S.; Li, X.; Chen, L.; Taylor, K.D.; Rotter, J.I.; Rissman, R.A.; Aisen, P.S.; Apostolova, L.G. Deleterious Effect of Butyrylcholinesterase K-Variant in Donepezil Treatment of Mild Cognitive Impairment. J. Alzheimers Dis. 2017, 56, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Martinelli-Boneschi, F.; Giacalone, G.; Magnani, G.; Biella, G.; Coppi, E.; Santangelo, R.; Brambilla, P.; Esposito, F.; Lupoli, S.; Clerici, F.; et al. Pharmacogenomics in Alzheimer’s disease: A genome-wide association study of response to cholinesterase inhibitors. Neurobiol. Aging 2013, 34, 1711.e7–1711.e13. [Google Scholar] [CrossRef] [PubMed]
- Becquemont, L.; Lecoeur, S.; Simon, T.; Beaune, P.; Funck-Brentano, C.; Jaillon, P. Glutathione S-transferase theta genetic polymorphism might influence tacrine hepatotoxicity in Alzheimer’s patients. Pharmacogenetics 1997, 7, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Torrellas, C.; López-Muñoz, F. Epigenomics of Alzheimer’s disease. J. Exp. Clin. Med. 2014, 6, 75–82. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Epigenetic drug discovery for Alzheimer’s disease. Expert Opin. Drug Discov. 2014, 9, 1059–1086. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.G.; Kim, R.B.; Wood, A.J.; Stein, C.M. Molecular basis of ethnic differences in drug disposition and response. Annu. Rev. Pharm. Toxicol. 2001, 41, 815–850. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. (Ed.) World Guide for Drug Use and Pharmacogenomics; EuroEspes Publishing Co.: La Coruña, Spain, 2012. [Google Scholar]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. [Google Scholar] [PubMed]
- Cacabelos, R. Pharmacogenomics of central nervous system (CNS) drugs. Drug Dev. Res. 2012, 73, 461–476. [Google Scholar] [CrossRef]
- Carril, J.C.; Cacabelos, R. Genomics and Pharmacogenomics of cerebrovascular disorders. J. Genom. Med. Pharmacogenom. 2016, 1, 27–55. [Google Scholar]
- Zhao, X.-S.; Peng, J.; Wu, Q.; Zhong, R.; Pan, L.-H.; Tang, Z.-H.; Jiang, Z.-S.; Wang, G.-X.; Liu, L.-S. Imbalanced cholesterol metabolism in Alzheimer’s disease. Clin. Chim. Acta 2016, 456, 107–114. [Google Scholar]
- Chen, Y.L.; Wang, L.M.; Chen, Y.; Gao, J.Y.; Marshall, C.; Cai, Z.Y.; Hu, G.; Xiao, M. Changes in astrocyte functional markers and β-amyloid metabolism-related proteins in the early stages of hypercholesterolemia. Neuroscience 2016, 316, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Dias, H.K.; Brown, C.L.; Polidori, M.C.; Lip, G.Y.; Griffiths, H.R. LDL-lipids from patients with hypercholesterolaemia and Alzheimer’s disease are inflammatory to microvascular endothelial cells: Mitigation by statin intervention. Clin. Sci. 2015, 129, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic Risk Factors of Sporadic Alzheimer’s Disease: Implications in the Pathology, Pathogenesis and Treatment. Aging Dis. 2015, 6, 282–299. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, F.F.; Chen, E.S.; Smith, M.C.; Bertolucci, P.H.F. Pharmacogenetics of angiotensin-converting enzyme inhibitors in patients with Alzheimer’s disease dementia. Curr. Alzheimer Res. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J.P.; Scutt, P.; Sprigg, N.; Bath, P.M. Hypercholesterolaemia and vascular dementia. Clin. Sci. 2017, 131, 1561–1578. [Google Scholar] [CrossRef] [PubMed]
- Sallustio, F.; Studer, V. Targeting New Pharmacological Approaches for Alzheimer’s Disease: Potential for Statins and Phosphodiesterase Inhibitors. CNS Neurol. Disord. Drug Targets 2016, 15, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Sinyavskaya, L.; Gauthier, S.; Renoux, C.; Dell’Aniello, S.; Suissa, S.; Brassard, P. Comparative effect of statins on the risk of incident Alzheimer disease. Neurology 2017. [Google Scholar] [CrossRef] [PubMed]
- Daneschvar, H.L.; Aronson, M.D.; Smetana, G.W. Do statins prevent Alzheimer’s disease? A narrative review. Eur. J. Intern. Med. 2015, 26, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Butterfield, D.A. Statins more than cholesterol lowering agents in Alzheimer disease: Their pleiotropic functions as potential therapeutic targets. Biochem. Pharmacol. 2014, 88, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Tamaoka, A. Dyslipidemia and Dementia. Brain Nerve 2016, 68, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E.; Royea, J.; Ongali, B.; Tong, X.K. Neurovascular and Cognitive failure in Alzheimer’s Disease: Benefits of Cardiovascular Therapy. Cell. Mol. Neurobiol. 2016, 36, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Samaras, K.; Brodaty, H.; Sachdev, P.S. Does statin use cause memory decline in the elderly? Trends Cardiovasc. Med. 2016, 26, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Zissimopoulos, J.M.; Barthold, D.; Brinton, R.D.; Joyce, G. Sex and Race Differences in the Association between Statin Use and the Incidence of Alzheimer Disease. JAMA Neurol. 2016, 74, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Y.; Shi, X.; Ma, C. An overview on therapeutics attenuating amyloid β level in Alzheimer’s disease: Targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am. J. Transl. Res. 2016, 8, 246–269. [Google Scholar] [PubMed]
- Buxbaum, J.D.; Cullen, E.I.; Friedhoff, L.T. Pharmacological concentrations of the HMGCoA reductase inhibitor lovastatin decrease the formation of the Alzheimer beta-amyloid peptide in vitro and in patients. Front. Biosci. 2002, 7, a50–a59. [Google Scholar] [PubMed]
- Yamamoto, N.; Fujii, Y.; Kasahara, R.; Tanida, M.; Ohora, K.; Ono, Y.; Suzuki, K.; Sobue, K. Simvastatin and atorvastatin facilitates amyloid β-protein degradation in extracellular spaces by increasing neprilysin secretion from astrocytes through activation of MAPK/Erk1/2 pathways. Glia 2016, 64, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Carril, J.C.; Teijido, O. Pharmacogenomics and epigenomics of age-related neurodegenerative disorders: Strategies for drug development. In Anti-Aging Drugs: From Basic Research to Clinical Practice; Vaiserman, A.M., Ed.; RSC Publishing: London, UK, 2017; Volume 57, pp. 75–141. [Google Scholar]
- Cacabelos, R.; Vallejo, A.I.; Lombardi, V.; Fernández-Novoa, L.; Pichel, V. E-SAR-94010 (LipoEsar®): A pleiotropic lipoprotein compound with powerful anti-atheromatous and lipid lowering effects. CNS Drug Rev. 2004, 10, 200–201. [Google Scholar] [CrossRef]
- Kitzmiller, J.P.; Mikulik, E.B.; Dauki, A.M.; Murkherjee, C.; Luzum, J.A. Pharmacogenomics of statins: Understanding susceptibility to adverse effects. Pharmacogenom. Personal. Med. 2016, 9, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M.; Mangravite, L.M.; Smith, J.D.; Medina, M.W.; Wang, D.; Guo, X.; Rieder, M.J.; Simon, J.A.; Hulley, S.B.; Waters, D.; et al. Variation in the 3-hydroxyl-3-methylglutaryl coenzyme a reductase gene is associated with racial differences in low-density lipoprotein cholesterol response to simvastatin treatment. Circulation 2008, 117, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Leduc, V.; Bourque, L.; Poirier, J.; Dufour, R. Role of rs3846662 and HMGCR alternative splicing in statin efficacy and baseline lipid levels in familial hypercholesterolemia. Pharmacogenet. Genom. 2016, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.J.; Rodin, A.S.; Klungel, O.H.; van Duijn, C.M.; Stricker, B.H.; van’t Slot, R.; de Boer, A.; Maitland-van der Zee, A.H. Pharmacogenetic interactions between ABCB1 and SLCO1B1 tagging SNPs and the effectiveness of statins in the prevention of myocardial infarction. Pharmacogenomics 2010, 11, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, J.; Kurkinen, K.; Neuvonen, P.; Niemi, M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin. Pharmacol. Ther. 2008, 84, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Guo, Y.; Wrughton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenom. J. 2011, 11, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Westlind-Johnsson, A.; Malmebo, S.; Johansson, A.; Otter, C.; Andersson, T.B.; Johansson, I.; Edwards, R.J.; Boobis, A.R.; Ingelman-Sundberg, M. Comparative analysis of CYP3A4 expression in human liver suggests only a minor role for CYP3A5 in drug metabolism. Drug Metab. Dispos. 2003, 31, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Park, P.W.; Lee, O.J.; Kang, D.K.; Park, J.Y. Effect of polymorphic CYP3A5 genotype on the single-dose simvastatin pharmacokinetics in healthy subjects. J. Clin. Pharmacol. 2007, 47, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Moonga, I.; Niccolini, F.; Wilson, H.; Pagano, G.; Politis, M. Alzheimer’s Disease Neuroimaging Initiative. Hypertension is associated with worse cognitive function and hippocampal hypometabolism in Alzheimer’s disease. Eur. J. Neurol. 2017, 24, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Wain, L.V.; Vaez, A.; Jansen, R.; Joehanes, R.; van der Most, P.J.; Erzurumluoglu, A.M.; O’Reilly, P.F.; Cabrera, C.P.; Warren, H.R.; Rose, L.M.; et al. Novel Blood Pressure Locus and Gene Discovery Using Genome-Wide Association Study and Expression Data Sets From Blood and the Kidney. Hypertension 2017, 70, e4–e19. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.D.; Ramirez, J.; Callahan, B.L.; Tobe, S.W.; Oh, P.; Berezuk, C.; Lanctôt, K.; Swardfager, W.; Nestor, S.; Kiss, A.; et al. Alzheimer’s Disease Neuroimaging Initiative. Antihypertensive Treatment is associated with MRI-Derived Markers of Neurodegeneration and Impaired Cognition: A Propensity-Weighted Cohort Study. J. Alzheimers Dis. 2017, 59, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Goel, R.; Shukla, S.; Shukla, R.; Hanif, K. Angiotensin Receptor Blockade by Inhibiting Glial Activation Promotes Hippocampal Neurogenesis via Activation of Wnt/β-Catenin Signaling in Hypertension. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sundbøll, J.; Hováth-Puhó, E.; Adelborg, K.; Schmidt, M.; Pedersen, L.; Bøtker, H.E.; Henderson, V.W.; Sørensen, H.T. Higher Risk of Vascular Dementia in Myocardial Infarction Survivors. Circulation 2017. [Google Scholar] [CrossRef] [PubMed]
- Schievink, S.H.J.; van Boxtel, M.P.J.; Deckers, K.; van Oostenbrugge, R.J.; Verhey, F.R.J.; Köhler, S. Cognitive changes in prevalent and incident cardiovascular disease: A 12-year follow-up in the Maastricht Aging Study (MAAS). Eur. Heart J. 2017, ehx365. [Google Scholar] [CrossRef] [PubMed]
- Gabin, J.M.; Tambs, K.; Saltvedt, I.; Sund, E.; Holmen, J. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: The HUNT Study. Alzheimers Res. Ther. 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Kern, K.C.; Wright, C.B.; Bergfield, K.L.; Fitzhugh, M.C.; Chen, K.; Moeller, J.R.; Nabizadeh, N.; Elkind, M.S.V.; Sacco, R.L.; Stern, Y.; et al. Blood Pressure Control in Aging Predicts Cerebral Atrophy Related to Small-Vessel White Matter Lesions. Front. Aging Neurosci. 2017, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Epigenomic networking in drug development: From pathogenic mechanisms to pharmacogenomics. Drug Dev. Res. 2014, 75, 348–365. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HMG CoA Reductase Inhibitors | ||
|---|---|---|
| Drug | Properties | Pharmacogenetics |
 | Name: ATORVASTATIN CALCIUM Molecular Formula: C66H68CaF2N4O10 Molecular Weight: 1155.341726 g/mol Mechanism: Inhibits HMG-CoA reductase, resulting in a compensatory increase in the expression of LDL receptors on hepatocyte membranes and a stimulation of LDL catabolism. Effect: Anticholesteremic Agent; HMG-CoA Reductase Inhibition; Apolipoprotein B reduction; Triglyceride reduction; Anti- atherosclerotic; Heart-health effects. | Pathogenic genes: ABCA1, ACE, APOA1, APOA5, APOB, APOC3, APOE, CETP, FGB, GNB3, LDLR, LIPC, MMP3, MTTP, NOS3, PON1 Mechanistic genes: ABCB1, ABCC1, APOA1, APOA5, APOB, APOC3, APOE, CRP, CYP11B2, HMGCR, IL10, IL6, LDLR, MMP3, PON1, TNF Metabolic genes:
Pleiotropic genes: APOA1, APOE, CRP, CYP11B2, ESR1, FGB, GNB3, HTR3B, IL6, IL10, ITGB3, MMP3, NOS3, TNF, USP5 |
 | Name: FLUVASTATIN SODIUM Molecular Formula: C24H25FNNaO4 Molecular Weight: 433.447772 g/mol Mechanism: Acts by competitively inhibiting HMGCR, the enzyme that catalyzes reduction of HMG-CoA to mevalonate. HDL is increased while total, LDL and VLDL cholesterols, apolipoprotein B, and plasma triglycerides are decreased. Effect: Anticholesteremic Agent; HMG-CoA Reductase Inhibition; Heart-health effects; Antineoplastic activity; Immune response modulation | Pathogenic genes: ABCA1, ACE, APOA1, APOA5, APOB, APOC3, APOE, CETP, CYP7A1, LDLR, LIPC, LPL, NOS3, PPARD, PON1 Mechanistic genes: APOA1, APOB, HMGCR, LPL, PON1 Metabolic genes:
Pleiotropic genes: ACE, APOA1, APOE, NOS3, NR1I2, NR1I3, PPARD, USP5 |
 | Name: LOVASTATIN Molecular Formula: C24H36O5 Molecular Weight: 404.53964 g/mol Mechanism: Acts by competitively inhibiting HMG-CoA reductase, enzyme which catalyzes rate-limiting step in cholesterol biosynthesis. Effect: Anticholesteremic Agent; HMG-CoA Reductase Inhibition; Heart-health effects; Antineoplastic activity. | Pathogenic genes: ABCA1, APOA1, APOA5, APOB, APOC3, CETP, LDLR, LIPC, LPL Mechanistic genes: APOA1, APOB, CETP, HMGCR, LDLR Metabolic genes:
Pleiotropic genes: TP53, USP5 |
 | Name: PITAVASTATIN Molecular Formula: C25H24FNO4 Molecular Weight: 421.460763 g/mol Mechanism: Works to control the synthesis of cholesterol via competitive inhibition of the liver enzyme, HMG-CoA reductase. As a result, a compensatory increase in LDL-receptor expression can be observed which facilitates an increase LDL catabolism. Effect: Anticholesteremic Agent; HMG-CoA Reductase Inhibition; Heart-health effects. | Pathogenic genes: APOB, LDLR Mechanistic genes: APOB, HMGC, LDLR, PPARG, PON1, VCAM1 Metabolic genes:
Pleiotropic genes: PPARG, VCAM1 |
 | Name: PRAVASTATIN SODIUM Molecular Formula: C23H35NaO7 Molecular Weight: 446.509569 g/mol Mechanism: A competitive inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. Effect: Anticholesteremic Agent; HMG-CoA Reductase Inhibition; Heart-health effects; Immune response modulation; MHC II suppression. | Pathogenic genes: ABCA1, ACE, APOA1, APOA5, APOB, APOC3, APOE, CETP, CYP7A1, FGB, LDLR, LIPC, LPL, NOS3 Mechanistic genes: APOA1, APOB, APOC3, APOE, CRP, HMGCR, LDLR, IL1B, IL6, IL10, MMP2, NOS3 Metabolic genes:
Pleiotropic genes: ACE, ALDH1A1, APOE, CBS, FGB, HTR3B, IL6, IL10, ITGB3, LEP, MTHFR, MMP2, MMP3, NOS3, TP53, USP5 |
 | Name: ROSUVASTATIN CALCIUM Molecular Formula: C44H54CaF2N6O12S2 Molecular Weight: 1001.137366 g/mol Mechanism: Inhibitor of HMG-CoA reductase, rate-limiting enzyme in cholesterol synthesis. This results in compensatory increase in expression of LDL receptors on hepatocyte membranes and stimulation of LDL catabolism. Effect: Anticholesteremic Agent; HMG-CoA Reductase Inhibition; Heart-health effects; Antineoplastic activity. | Pathogenic genes: ABCA1, ACE, APOA1, APOA5, APOB, APOC3, APOE, CETP CYP7A1, FGB, LDLR, LIPC, LPL, NOS3 Mechanistic genes: APOA1, APOB, CETP, FGB, HMGCR, LDLR, LPL, NOS3 Metabolic genes:
Pleiotropic genes: ACE, APOE, FGB, ITGB3, NOS3, TCF20, USP5 |
 | Name: SIMVASTATIN Molecular Formula: C25H38O5 Molecular Weight: 418.56622 g/mol Mechanism: Prodrug requiring hydrolysis in vivo for activity. Inhibits HMG-CoA reductase, causing subsequent reduction in hepatic cholesterol synthesis. Reduces serum concentrations of total cholesterol, LDL-C, Apo B, and triglycerides Effect: Anticholesteremic Agent; HMG-CoA Reductase Inhibition; Heart-health effects; Anti-inflammatory activity | Pathogenic genes: ABCA1, APOA1, APOA5, APOB, APOC3, APOE, CETP, CYP7A1, FGB, GNB3, LIPC, LDLR, LPL, NOS3 Mechanistic genes: ABCA1, APOA1, APOB, APOE, CETP, HMGCR, IL6, LDLR, LPL, VCAM1 Metabolic genes:
Pleiotropic genes: APOE, F2, FGB, GNB3, NOS3, PRNP, TNF, VCAM1, USP5 |
| Hypotensive Agents | ||
|---|---|---|
| Renin-Angiotensin-Aldosterone System Inhibitors | ||
| Angiotensin-Converting Enzyme Inhibitors | ||
| Drug | Properties | Pharmacogenetics |
 | Name: Benazepril Hydrochloride IUPAC Name: 1H-1-Benzazepine-1-acetic acid, 3-[[1-(ethoxycarbonyl)-3-phenylpropyl]amino]-2,3,4,5-tetrahydro-2-oxo-, monohydrochloride, [S-(R*,R*)]- Molecular Formula: C24H28N2O5·HCl Molecular Weight: 460.95 g/mol Mechanism: Competitive inhibition of ACE activity, which regulates the conversion of angiotensin I to angiotensin II, a potent vasoconstrictor, with resultant lower levels of angiotensin II which causes an increase in plasma renin activity and a reduction in aldosterone secretion. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, ACE2, ADD1, ADRB2, AGT, AGTR1, MTHFR, MTR Metabolic genes Substrate: CYP11B2 Transporter genes: SLC15A1, SLC15A2 |
 | Name: Captopril IUPAC Name: l-Proline, 1-[(2S)-3-mercapto-2-methyl-1-oxopropyl]- Molecular Formula: C9H15NO3S Molecular Weight: 217.29 g/mol Mechanism: Competitive inhibitor of angiotensin-converting enzyme (ACE). Prevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor, resulting in lower levels of angiotensin II, which causes an increase in plasma renin activity and a reduction in aldosterone secretion. By decreasing local angiotensin II production, ACE inhibitors may decrease vascular tone by reducing direct angiotensin II-induced vasoconstriction and/or angiotensin II-induced increases in sympathetic activity. In hypertensive patients, captopril reduces blood pressure by decreasing total peripheral resistance with no change or an increase in heart rate, stroke volume, or cardiac output (these effects are independent of pre-treatment blood pressure/cardiac output). Causes arterial and possibly venous dilation. In patients with congestive heart failure, captopril decreases total peripheral resistance, pulmonary vascular resistance, pulmonary capillary wedge pressure, and mean arterial and right atrial pressures (cardiac index, cardiac output, stroke volume, and exercise tolerance are increased; heart rate decreases or is unchanged). The drug may also cause regional redistribution of blood flow, principally increasing renal blood flow (glomerular filtration rate is usually unchanged) with slight or no increase in flow in the forearm or hepatic vasculature, respectively. The hypotensive effect of captopril persists longer than inhibition of ACE in blood (unknown whether ACE is inhibited longer in vascular endothelium than in blood). Captopril alone is apparently more effective in reducing blood pressure in high or normal renin hypertension. Serum prolactin concentration has been reported to increase during captopril therapy. Effect: Angiotensin-Converting Enzyme Inhibition. Antihypertensive Agent. | Mechanistic genes: ACE, ACE2, AGT, ALB, BDKRB2, CHRNA2, LTA4H, MMP2, MMP9, NOS3, REN Metabolic genes Substrate: CYP2D6, CYP3A4, CYP3A5, CYP11B2 Transporter genes: ABCB1, SLC15A1, SLC22A6 |
 | Name: Cilazapril IUPAC Name: 6H-Pyridazino[1,2-a][1,2]diazepine-1-carboxylic acid, 9-[[1-(ethoxycarbonyl)-3-phenylpropyl]amino]octahydro-10-oxo-, monohydrate, [1S-[1α,9α(R*)]]- Molecular Formula: C22H31N3O5·H2O Molecular Weight: 435.51 g/mol Mechanism: Competitive inhibitor of angiotensin-converting enzyme. Prevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor, resulting in lower levels of angiotensin II, and thus causing an increase in plasma renin activity and a reduction in aldosterone secretion. Effect: Renin-Angiotensin-Aldosterone System Inhibition; Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE Transporter genes: ABCB1, SLC15A1, SLC15A2 |
 | Name: Enalapril Maleate IUPAC Name: l-Proline, 1-[N-[1-(ethoxycarbonyl)-3-phenylpropyl]-l-alanyl]-, (S)-, (Z)-2-butenedioate (1:1) Molecular Formula: C20H28N2O5·C4H4O4 Molecular Weight: 492.52 g/mol Mechanism: Competitive inhibitor of angiotensin-converting enzyme (ACE). Prevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor. Results in lower levels of angiotensin II, which causes an increase in plasma renin activity and a reduction in aldosterone secretion. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, ADRB2, AGT, AGTR1, BDKRB2, NOS3 Metabolic genes Substrate: CYP3A4, CYP3A5 Transporter genes: SLC15A1, SLC22A6, SLC22A7, SLC22A8, SLCO1A2 Pleiotropic genes: IL6 |
 | Name: Fosinopril Sodium IUPAC Name: l-Proline, 4-cyclohexyl-1-[[[2-methyl-1-(1-oxopropoxy)propoxy](4-phenylbutyl)phosphinyl]acetyl]-, sodium salt, [1[S*(R*)],2a,4b]- Molecular Formula: C30H45NNaO7P Molecular Weight: 585.64 g/mol Mechanism: Competitive inhibitor of angiotensin-converting enzyme. Prevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor. Results in lower levels of angiotensin II, which causes increase in plasma renin activity and reduction in aldosterone secretion. Vasoactive kallikreins may be decreased in conversion to active hormones by ACEIs, thus reducing blood pressure. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, AGT, AGTR1, BDKRB2, NOS3, PDE1C Transporter genes: SLC15A1, SLC15A2 |
 | Name: Lisinopril IUPAC Name: l-Proline, 1-[N2-(1-carboxy-3-phenylpropyl)-l-lysyl]-, dihydrate, (S)- Molecular Formula: C21H31N3O5·2H2O Molecular Weight: g/mol Mechanism: Competitive inhibitor of angiotensin-converting enzyme (ACE). Prevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, ACE2, ADD1, AGT, AGTR1, BDKRB2, MMP3, NOS3, NPPA Metabolic genes Substrate: CYP3A4, CYP3A5 Transporter genes: ABCB1 |
 | Name: Moexipril Hydrochloride IUPAC Name: 3-Isoquinolinecarboxylic acid, 2-[2-[[1-(ethoxycarbonyl)-3-phenylpropyl]amino]-1-oxopropyl]-1,2,3,4-tetrahydro-6,7-dimethoxy-, monohydrochloride, [3S-[2[R*(R*)],3R*]]- Molecular Formula: C27H34N2O7·HCl Molecular Weight: 535.03 g/mol Mechanism: Prevents conversion of angiotensin I to angiotensin II, potent vasoconstrictor, resulting in lower levels of angiotensin II; this causes increase in plasma renin activity and reduction in aldosterone secretion. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, ACE2, AGT Metabolic genes Substrate: CES1 Transporter genes: SLC15A1, SLC15A2 |
 | Name: Perindopril Erbumine IUPAC Name: 1H-Indole-2-carboxylic acid, 1-[2-[[1-(ethoxycarbonyl)butyl]amino]-1-oxopropyl]octahydro-, [2S-[1[R*(R*)],2α,3aβ,7aβ]]-, compd. with 2-methyl-2-propanamine (1:1) Molecular Formula: C19H32N2O5·C4H11N Molecular Weight: 441.60 g/mol Mechanism: A prodrug for perindoprilat, which acts as competitive inhibitor of angiotensin-converting enzyme. Prevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor, and causes increase in plasma renin activity and reduction in aldosterone secretion. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, AGT, AGTR1, BCHE, MMP2, TGFB1, SFRP4 Transporter genes: ABCB1, SLC15A1, SLC15A2 |
 | Name: Quinapril Hydrochloride IUPAC Name: 3-Isoquinolinecarboxylic acid, 2-[2-[[1-(ethoxycarbonyl)-3-phenylpropyl]amino]-1-oxopropyl]-1,2,3,4-tetrahydro-, monohydrochloride, [3S-[2[R*(R*)],3R*]] Molecular Formula: C25H30N2O5 Molecular Weight: 474.98 g/mol Mechanism: Prodrug, not pharmacologically active until hydrolyzed in liver to quinaprilat. Competitive inhibitor of ACE. Prevents conversion of angiotensin I to angiotensin II, a potent vasoconstrictor. Lower levels of angiotensin II cause increase in plasma renin activity and reduction in aldosterone secretion. A CNS mechanism may also be involved in hypotensive effect as angiotensin II increases adrenergic outflow from CNS. Vasoactive kallikreins may be decreased in conversion to active hormones by ACEIs, thus reducing blood pressure. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, AGT, AGTR1, BDKBR2, NR1I2, TGFB1 Metabolic genes Substrate: CYP11B2 Transporter genes: SLC15A1, SLC15A2 |
 | Name: Ramipril IUPAC Name: Cyclopenta[b]pyrrole-2-carboxylic acid, 1-[2-[[1-(ethoxycarbonyl)-3-phenylpropyl]amino]-1-oxopropyl]octahydro-, [2S-[1[R*(R*)],2α,3aβ,6ab]]- Molecular Formula: C23H32N2O5 Molecular Weight: 416.51 g/mol Mechanism: Ramipril undergoes enzymatic saponification by esterases in liver, to its active metabolite ramiprilat. Ramiprilat reversibly binds to angiotensin-converting enzyme, thus preventing formation of potent vasoconstrictor angiotensin II from angiotensin I. CNS mechanism may also be involved in hypotensive effect, as angiotensin II increases adrenergic outflow from CNS. Vasoactive kallikreins may be decreased in conversion to active hormones by ACEIs, thus reducing blood pressure. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, AGT, AGTR1, BCHE, BDKRB2, COL1A1, MMP2, NOS3, REN, TGBFB1 Metabolic genes Substrate: CYP11B2 Pleiotropic genes: APOE Transporter genes: SLC15A1, SLC15A2 |
 | Name: Trandolapril IUPAC Name: (2S,3aR,7aS)-1-[(S)-N-[(S)-1-Carboxy-3-phenylpropyl]alanyl]hexahydro-2-indolinecarboxylic acid, 1-ethyl ester Molecular Formula: C24H34N2O5 Molecular Weight: 430.54 g/mol Mechanism: Prevents formation of angiotensin II from angiotensin I. Trandolapril must undergo enzymatic hydrolysis, mainly in liver, to its biologically active metabolite, trandolaprilat. A CNS mechanism may also be involved in hypotensive effect as angiotensin II increases adrenergic outflow from CNS. Vasoactive kallikreins may be decreased in conversion to active hormones by ACEIs, thus reducing blood pressure. Effect: Angiotensin-Converting Enzyme Inhibition. | Mechanistic genes: ACE, ADD1 Metabolic genes Substrate: CES1 Transporter genes: SLC15A1, SLC15A2 |
| Angiotensin II Receptor Antagonists | ||
| Drug | Properties | Pharmacogenetics |
 | Name: Candesartan Cilexetil IUPAC Name: 1H-Benzimidazole-7-carboxylic acid, 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]- Molecular Formula: C24H20N6O3 Molecular Weight: 440.45 g/mol Mechanism: Candesartan is an angiotensin receptor antagonist, blocking vasoconstriction and the aldosterone-secreting effects (reabsorption of sodium and water) of angiotensin II. Effect: Angiotensin II Receptor Antagonists. | Mechanistic genes: ACE, AGT, AGTR1, BDKRB2, NOS3, PTGS1, TGFB1 Metabolic genes Substrate: CYP1A1, CYP2C8, CYP2C9, CYP11B2, UGT1A3, UGT1A5, UGT2B7 Transporter genes: ABCB1, ABCG2 |
 | Name: Eprosartan Mesylate IUPAC Name: 2-Thiophenepropanoic acid, α-[[2-butyl-1-[(4-carboxyphenyl)methyl]-1H-imidazol-5-yl]methylene]-, (E)-, monomethanesulfonate Molecular Formula: C23H24N2O4S·CH4O3S Molecular Weight: 520.62 g/mol Mechanism: A nonbiphenyl, nontetrazole angiotensin II receptor (AT1) antagonist. Blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor in many tissues, such as vascular smooth muscle and adrenal gland. Does not bind to or block other hormone receptors or ion channels known to be important in cardiovascular regulation. Effect: Angiotensin II Receptor Antagonists. | Mechanistic genes: ACE, AGTR1 Metabolic genes Substrate: CYP2C9 Transporter genes: ABCB1, ABCC2, ABCG2 |
 | Name: Irbesartan IUPAC Name: 1,3-Diazaspiro[4.4]non-1-en-4-one, 2-butyl-3-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]- Molecular Formula: C25H28N6O Molecular Weight: 428.53 g/mol Mechanism: Irbesartan binds to AT1 angiotensin II receptor. This binding prevents angiotensin II from binding to receptor, thereby blocking the vasoconstriction and aldosterone-secreting effects of angiotensin II. Effect: Angiotensin II Receptor Antagonists. | Mechanistic genes: ADRA1A, AGT, AGTR1, APOB, BDKRB2, JUN, LDLR, NOS3, PTGS1, TGFB1 Metabolic genes Substrate: CYP1A2, CYP2C8, CYP2C9, CYP2D6, CYP3A4, CYP3A5, CYP11B2, UGT1A3 Transporter genes: ABCB1 ABCG2 Pleiotropic genes: APOE |
 | Name: Losartan Potassium IUPAC Name: 1H-Imidazole-5-methanol, 2-butyl-4-chloro-1-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]-, monopotassium salt Molecular Formula: C22H22ClKN6O Molecular Weight: 461.00 g/mol Mechanism: As a selective and competitive nonpeptide angiotensin II receptor antagonist, losartan blocks vasoconstrictor and aldosterone-secreting effects of angiotensin II. Losartan increases urinary flow rate and, in addition to being natriuretic and kaliuretic, increases excretion of chloride, magnesium, uric acid, calcium, and phosphate. Effect: Angiotensin II Receptor Antagonists. | Mechanistic genes: ACE, ADD1, AGT, AGTR1, AGTR2, ALB, BDKRB2, EDN1, FOS, MMP2, NOS3, PDGFRB, TGFB1 Metabolic genes Substrate: CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP3A4, CYP3A5, CYP11B2, UGT1A1, UGT1A3, UGT1A10, UGT2B7, UGT2B17 Transporter genes: ABCB1, ABCG2, SLC2A9, SLC22A6, SLC22A12 Pleiotropic genes: TNF |
 | Name: Olmesartan Medoxomil IUPAC Name: 1H-Imidazole-5-carboxylic acid, 4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2′-(1H-tetrazol-5-yl) [1,1′-biphenyl]-4-yl]methyl]-, (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl ester Molecular Formula: C29H30N6O6 Molecular Weight: 558.59 g/mol Mechanism: Blocks vasoconstrictor and aldosterone-secreting effects of angiotensin II. Interacts reversibly at AT1 and AT2 receptors and has slow dissociation kinetics (has greater affinity for AT1 receptor). Olmesartan increases urinary flow rate and, besides being natriuretic and kaliuretic, increases excretion of chloride, magnesium, uric acid, calcium, and phosphate. Effect: Angiotensin II Receptor Antagonists. | Mechanistic genes: AGTR1, ACE2, EDN1, TGFB1 Metabolic genes Substrate: CMBL, CYP2C9 Transporter genes: ABCB1, ABCC2, ABCG2, SLCO1B1, SLCO1B3, SLCO1A2, SLC22A8 Pleiotropic genes: APOE |
 | Name: Telmisartan IUPAC Name: [1,1′-Biphenyl]-2-carboxylic acid, 4′-[(1,4′-dimethyl-2′-propyl[2,6′-bi-1H-benzimidazol]-1′-yl)methyl]- Molecular Formula: C33H30N4O2 Molecular Weight: 514.62 g/mol Mechanism: A nonpeptide AT1 angiotensin II receptor antagonist. This binding prevents angiotensin II from binding to receptor thereby blocking vasoconstriction and aldosterone-secreting effects of angiotensin II. Effect: Angiotensin II Receptor Antagonists. | Mechanistic genes: ACE, AGT, AGTR1, BDKRB2, ERAP1, PPARG Metabolic genes Substrate: CYP2C9, CYP2C19, CYP11B2, UGT1A1 Transporter genes: ABCB1, ABCC2, ABCG2 |
 | Name: Valsartan IUPAC Name: l-Valine, N-(1-oxopentyl)-N-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]- Molecular Formula: C24H29N5O3 Molecular Weight: 435.52 g/mol Mechanism: Displaces angiotensin II from AT1 receptor and produces its blood pressure-lowering effects by antagonizing AT1-induced vasoconstriction, aldosterone release, catecholamine release, arginine vasopressin release, water intake, and hypertrophic responses. Effect: Angiotensin II Receptor Antagonists. | Mechanistic genes: ACE, AGT, AGTR1, ALB, BDKRB2, ERAP1, GNB3, Metabolic genes Substrate: CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5, CYP11B2 Transporter genes: ABCC2, SLCO1B1, SLCO1B3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacabelos, R.; Meyyazhagan, A.; Carril, J.C.; Cacabelos, P.; Teijido, Ó. Pharmacogenetics of Vascular Risk Factors in Alzheimer’s Disease. J. Pers. Med. 2018, 8, 3. https://doi.org/10.3390/jpm8010003
Cacabelos R, Meyyazhagan A, Carril JC, Cacabelos P, Teijido Ó. Pharmacogenetics of Vascular Risk Factors in Alzheimer’s Disease. Journal of Personalized Medicine. 2018; 8(1):3. https://doi.org/10.3390/jpm8010003
Chicago/Turabian StyleCacabelos, Ramón, Arun Meyyazhagan, Juan C. Carril, Pablo Cacabelos, and Óscar Teijido. 2018. "Pharmacogenetics of Vascular Risk Factors in Alzheimer’s Disease" Journal of Personalized Medicine 8, no. 1: 3. https://doi.org/10.3390/jpm8010003
APA StyleCacabelos, R., Meyyazhagan, A., Carril, J. C., Cacabelos, P., & Teijido, Ó. (2018). Pharmacogenetics of Vascular Risk Factors in Alzheimer’s Disease. Journal of Personalized Medicine, 8(1), 3. https://doi.org/10.3390/jpm8010003