Electrochemical Recycling of Platinum Group Metals from Spent Catalytic Converters
by

Cornelia Diac
1,2, 
Florentina Iuliana Maxim
1,2,
Radu Tirca
2,
Adrian Ciocanea
3,
Valeriu Filip
2,
Eugeniu Vasile
4,* and
Serban N. Stamatin
1,* 1
3Nano-SAE Research Centre, University of Bucharest, 077125 Bucharest, Romania
2
Faculty of Physics, University of Bucharest, 077125 Bucharest, Romania
3
Power Engineering Faculty; Hydraulics, Hydraulic Machines, and Environmental Engineering Department, University “Politehnica” of Bucharest, 060042 Bucharest, Romania
4
Department of Oxide Materials and Nanomaterials, Faculty of Applied Chemistry and Material Science, University “Politehnica” of Bucharest, 060042 Bucharest, Romania
*
Authors to whom correspondence should be addressed.
Metals 2020, 10(6), 822; https://doi.org/10.3390/met10060822
Submission received: 24 May 2020
/
Revised: 12 June 2020
/
Accepted: 16 June 2020
/
Published: 19 June 2020
(This article belongs to the Special Issue Metal Removal and Recycling)
Abstract
:Platinum group metals (PGMs: Pt, Pd, and Rh) are used extensively by the industry, while the natural resources are limited. The PGM concentration in spent catalytic converters is 100 times larger than in natural occurring ores. Traditional PGM methods use high temperature furnaces and strong oxidants, thus polluting the environment. Electrochemical studies showed that platinum can be converted to their chloride form. The amount of dissolved PGM was monitored by inductively coupled plasma-optical emission spectroscopy and the structure was identified by ultraviolet-visible spectroscopy. An electrochemistry protocol was designed to maximize platinum dissolution, which was then used for a spent catalytic converter. A key finding is the use of potential step that enhances the dissolution rate by a factor of 4. Recycling rates as high as 50% were achieved in 24 h without any pretreatment of the catalyst. The method developed herein is part of a current need to make the PGM recycling process more sustainable.
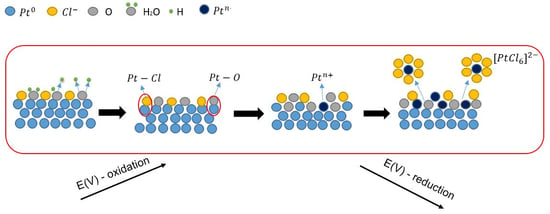
1. Introduction
Sustainable societies must strike a balance between economy, society, and the environment. Climate change, if left unaddressed, will have a negative impact on the environment that will cause large societal changes and threaten the current economic model. Metals are paramount in delivering sustainable solutions to tackle future challenges. More specifically, platinum group metals (PGMs) are used in catalysis, renewable energy, and pharmaceutical sectors, which are central to industrialized societies. The European Union (EU) released the list of Critical Raw Materials in order to bolster mining and recycling activities in line with circular economy principles and to assure the supply of crucial materials [1].
PGMs, such as platinum and palladium, are widely used in cars [2,3]. The PGM concentration in an automotive catalyst range between 0.1% and 0.2%, which is 100 times larger than a typical PGM mine (i.e., 0.001%). The sheer difference in concentrations between urban wastes and ores established a new concept, that is, urban mining [4]. Extracting PGMs from urban wastes without damaging the environment sits at the forefront of future recycling policies.
Conventional methods for recovering PGMs can be divided into pyro- and hydrometallurgical [4,5]. The pyrometallurgical process is based on the nickel or copper metallurgy and can be economically viable only at large scale owing to the requirements of high-temperature melting and chlorination at high temperatures as well as the initial investment. Hydrometallurgy uses highly oxidant reagents, such as aqua regia, to dissolve Pt, while also releasing considerable amounts of NOx gases [6]. Another possibility is the use of chlorine leaching in aqueous solutions [7]. Combining pyro- and hydrometallurgical processes has seen an increased interest recently [8]. Both methods are large generators of highly corrosive and toxic aqueous waste, which must be treated before discharge. In consequence, stricter environmental regulations are paving the way towards improved waste treatment and to developing alternative solutions.
Automotive catalysts lose approximately 25% of their initial efficiency in 160,000 km, which also represents their lifetime [9]. Autocatalysts are collected locally and then transferred to larger recycling facilities. There is a need to investigate solutions that have a reduced environmental footprint and can exploit local resources to achieve high recovery PGM rates. Excess renewable energy has been previously used in connection with water electrolyzers to store hydrogen [10] or to reduce greenhouse gases [11]. Similarly, electrochemical processes that treat wastes can use excess renewable energy to run a reaction at practically no energy cost. This can be part of regional policies to treat waste in general.
An alternative method to electrochemically recycle PGM from spent automotive catalysts without damaging the environment is investigated herein. PGMs are recycled by converting nanoparticles into aqueous metal chloro complexes. The extensive use of Pt in polymer electrolyte fuel cells revealed that chlorides at trace levels significantly degrade Pt’s electrochemical activity [12]. Pt dissolution know-how can be transferred to other PGMs. The dissolution behavior of Pt in chloride containing electrolytes at acidic and neutral pH is investigated. Sulphate-based electrolytes are used as dissolution background to stress the need of chlorides. The electrochemistry of Pt in chloride containing electrolytes at acidic and neutral pH is discussed initially. An electrochemical protocol is designed and tested on a spent automotive catalyst without pretreatment. The results show an approximately 50% PGM recovery efficiency in 24 h.
2. Materials and Methods
Sulphuric acid and potassium sulphate were purchased from Silal Trading (Romania), potassium chloride was purchased from Chimactiv (Romania), and hydrochloric acid was purchased from Lachner (Czech Republic). All chemicals were used without further purification. Ultrapure water (>18.2 MΩ*cm) was supplied by Milli-Q Direct Q System (Burlington, MA, USA). The electrochemical experiments were conducted in a three-electrode, two-compartment glass cell. The potentiostat (OrigaFlex—OGF500) (Lyon, France) was controlled by OrigaView. A polycrystalline platinum electrode was used as a working electrode. Saturated calomel (Hg/Hg2Cl2), Ag/AgCl, or Hg/Hg2SO4 electrodes were used as reference electrodes. Chloride-based reference electrodes were used for chloride-based electrolytes to avoid contamination of the electrolyte in the working cell. Similarly, Hg/Hg2SO4 was used for 0.5 M H2SO4 and 0.5 M K2SO4. A graphite rod was used as a counter electrode. The reference electrode potential was measured against a home-built standard H2 electrode (see Supplementary Materials). All potentials were converted to the reversible hydrogen electrode (RHE) scale using Equation S1 (see Supplementary Materials). The experiments were performed in four electrolyte solutions: two of them based on chlorine (1 M HCl, 1 M KCl) and two based on sulfate (0.5 M H2SO4, 0.5 M K2SO4). Electrolyte solutions were prepared from concentrated HCl (35%), H2SO4 (94–96%), KCl (99%), and K2SO4 (99%) diluted in ultrapure water (>18.2MΩ*cm). To keep a similar pH and ionic strength, we prepared 1 M HCl and 0.5 M H2SO4. We prepared 0.5 M H2SO4 and 0.5 M K2SO4 to maintain the same sulphate concentration. The cell was left overnight in boiling ultrapure water (80 °C) before every experiment. The duration of the experiments was 24 h unless otherwise mentioned. All measurements were performed at room temperature. A spent automotive catalyst was supplied by a local scrap metal shop. The average Pt mass determined by inductively coupled plasma-optical emission spectrometry (ICP-OES) was (0.135 ± 0.003) g Pt per kg of spent automotive catalyst, which means approximately 0.014% Pt, although not present in the X-ray diffraction (XRD). A Pd concentration of (0.458 ± 0.025) g Pd per kg was measured, corresponding to approximately 0.046% Pd.
ICP-OES (Perkin Elmer, Avio 200) (Waltham, MA, USA) was used to determine the amount of platinum dissolved. Further details on ICP-OES can be found in Supplementary Materials. UV/Vis (Jasco V550, Tokyo, Japan) was used to determine the presence of platinum complexes from solutions. Powder X-ray diffraction was measured by means of Panalytical X’PER PRO (Almelo, The Netherlands) equipped with a Cu Kα radiation source (λ = 1.54 Å) and an Ni filter. Morphological characterization was carried out on a FEI TECNAI F30 G2 S-TWIN (Hillsboro, OR, USA) high-resolution transmission electron microscope with a field emission gun at a maximum voltage of 300 kV equipped with an energy dispersive X-ray (EDX) detector. Samples were homogenized in an ultrasound bath for 15 min, and then a drop of solution was placed on a holey carbon-coated copper grid, followed by natural drying.
3. Results
3.1. Effect of pH and Anion on Platinum Electrochemistry
Figure 1 shows the first and last cyclic voltammogram (CV) in chloride (A and C) and sulphate (B and D) containing electrolytes at acidic pH and neutral pH. Anodic reactions, such as oxidation of molecular hydrogen, hydrogen desorption, platinum oxide formation (Table 1, R6), and oxygen (Table 1, R5) and chlorine evolution (Table 1, R4), are observed at positive currents. Cathodic reactions (hydronium/hydroxyl ion adsorption and reduction to hydrogen, oxygen reduction, and chlorine reduction) can be observed at negative currents. Reactions R1–3 (Table 1) cannot be clearly distinguished in the CVs of Figure 1. The potential window was tuned in such a way to cover the main reactions from Table 1.
The CVs showed the well-defined features known as the Pt electrochemical “fingerprint”, which can be divided into two regions [14,15]. The first region is presented in the insets of Figure 1 and is also known as the Pt-hydride region. The cathodic sweep (negative current) in the insets of Figure 1 shows the typical response for hydronium and/or hydroxyl adsorption, which are further reduced to hydrogen. Hydrogen evolution reaction at negative potentials in neutral pH is documented in the literature [16,17]. On the anodic sweep (positive current), molecular hydrogen is oxidized and desorbed. The second region (>0.8 V vs. RHE) shows the oxidation of Pt, followed by oxygen and/or chlorine evolution on the anodic sweep. On the cathodic sweep, oxygen is reduced to water together with chlorine reduction in chloride containing electrolytes at potentials larger than 1.2 V versus RHE. A more detailed electrochemical characterization is available in the Supplementary Materials.
Figure 2A shows the concentration of Pt found in the electrolyte determined by ICP-OES at the end of the electrochemical testing. The largest Pt concentration was found in HCl, followed by KCl and H2SO4. There was no Pt detected in K2SO4 (the reader should bear in mind that 0.01 ppm is the minimum detection limit). It is challenging to fully exclude the presence of Pt in K2SO4. There are two orders of magnitude difference between 1 M HCl (2.461 ppm) and 0.5 M H2SO4 (0.061 ppm). Similar to acidic electrolytes, there are at least two orders of magnitude between KCl (0.311 ppm) and K2SO4. Almost an order of magnitude difference is observed between HCl and KCl. The total dissolved Pt trend is in agreement with the loss in electrode roughness, that is, the largest loss in roughness correlates with the greatest dissolution.
Figure 2B shows the UV/vis spectra of the solutions. The samples in chloride electrolytes show a strong peak at 262 nm, which has been previously assigned to [PtCl6]2− [18]. Sulphate containing solutions do not show any identifiable feature as there is no known Pt-sulphate compound that can be synthesized [19].
3.2. Electrochemical Dissolution of Pt in Hydrochloric Acid
We have shown that the presence of chloride is mandatory to achieve high Pt dissolution rates. In the following, we explore mass transport and kinetic limitations on Pt dissolution rates. We chose the electrolyte in which Pt shows the largest dissolution rate in order to circumvent any issues related to the lower detection limits of the ICP-OES. The results can be transferred to other electrolytes.
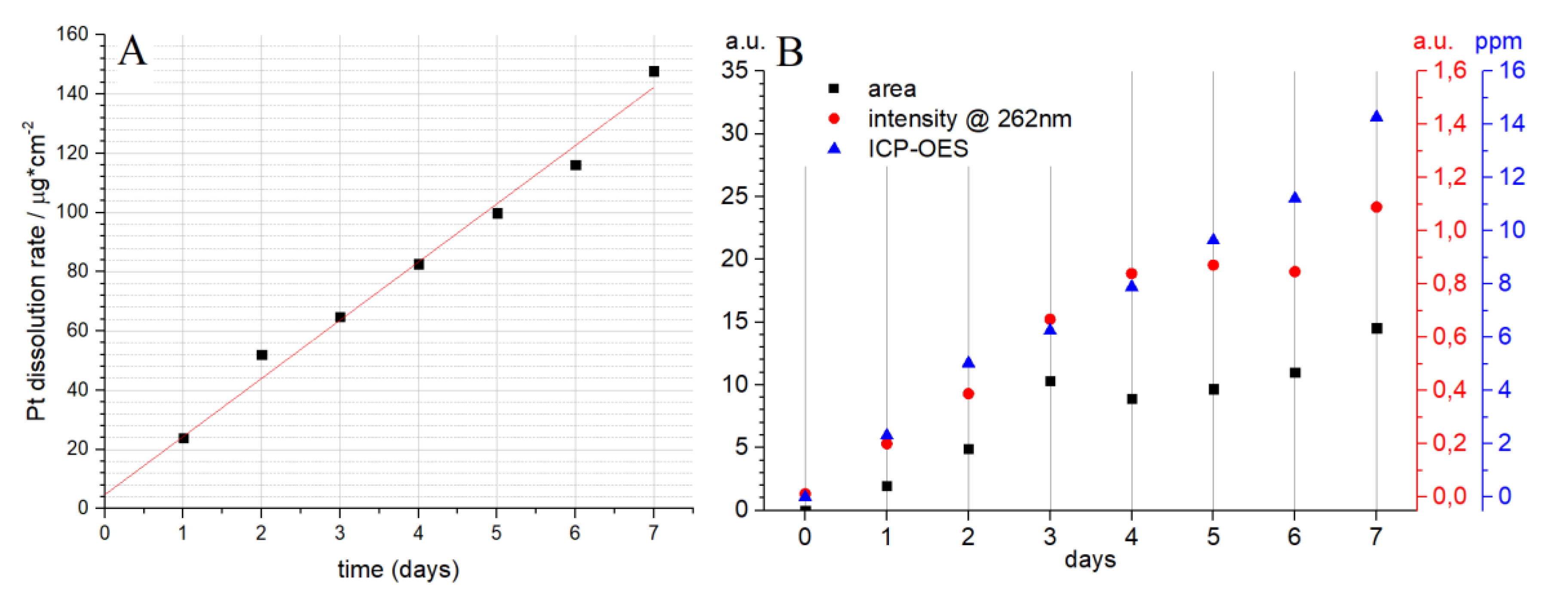
The amount of Pt electrochemically dissolved per day in 1 M HCl was determined by ICP-OES (Figure 3A). The amount of dissolved Pt increases linearly from day 1 to day 7. The intercept on the y-axis gives the amount of Pt at day 0, that is, 4.752 µgPt/cm2, which can be considered as 0. The slope of the fitted line gives the rate of Pt dissolution per day, that is, 19.64 µgPt/cm2 per day. The chemical structure of the compound was identified as [PtCl6]2− as resulting from UV/vis analysis (Figure 2B). The UV/vis spectrum at the beginning of the experiments (day 0) shows only a peak at 205 nm (black line in Figure S3, Supplementary Materials). Starting from day 1, there is an increase in the absorbance at 262 nm and the peak at 205 nm shifts to 215 nm. The absorption was determined by integrating the area under the peak and compared with the absorbance at 262 nm (Figure 3B). The Beer-Lambert law states that the absorbance is directly proportional to the concentration. There is a constant increase in the absorbance at 262 nm, with some deviation between days 4 to 6 (Figure 3B). A similar trend can be observed between days 3 and 6 if one measures the area of the peak (Figure 3B). Figure 3B shows that UV/vis can be used for the determination of [PtCl6]2−, but ICP-OES is a more desired technique owing to its considerably better accuracy and lower detection limit.
Pt dissolution follows a linear trend during 7 days (Figure 3A), suggesting that there are no mass transport limitations to and from the electrode. To test this hypothesis, Pt rotating disc electrode (A = 0.0314 cm2) was used to measure the Pt dissolution rate as a function of the rotation rate. A rotating disc electrode has a controlled mass transport regime that is governed only by the rotation rate of the electrode (see Supplementary Materials). Figure 4A shows the Pt dissolution as determined by ICP-OES at 0, 900, and 1600 rpm from a Pt rotating disc electrode (Figure 4A). The rotation rate does not affect the amount of dissolved Pt. There is a slight difference in the Pt dissolution rate at 0 rpm in Figure 4A and the CV in Figure 4B. The reader should bear in mind that the geometrical area of the Pt RDE (0.0314 cm−2) is 10-fold smaller than the static Pt electrode (0.322 cm−2) used for the experiments in Figure 4B, which can cause minor deviations.
3.3. Potential Sweep versus Potential Step
It has recently been established that the electrochemical dissolution of Pt is accelerated by transient conditions [20,21]. We made use of this concept to further accelerate Pt dissolution by exposing the surface to rapid changes in potential. Figure 4B shows the dissolution of Pt by four electrochemical procedures: CVs between 0.45 and 1.25 V versus RHE at a scan rate of 0.1 V s−1 (1); chronoamperometry-potential step (PS) experiments with a 3 s step at 0.45 V and 1.25 V versus RHE (2); chronoamperometry-potential hold experiment at 0.45 V versus RHE (3); and potential hold experiment at 1.25 V versus RHE (4). The time was kept constant in all the experiments.
The experiments in which Pt is exposed only to an oxidation potential (i.e., 1.25 V vs. RHE) show minor dissolution. A similar behavior can be observed at potentials where Pt is only reduced (i.e., 0.45 V vs. RHE). In experiments with potential changes of CV and PS (Figure 4B), they are 20.8 and 89.1 µg·h·cm−2geo, respectively.
3.4. Morphology and Composition of the Automotive Catalyst
There are several types of automotive catalysts that are currently in use [22,23]. The one used in this study has three mixed compounds: magnesium doped aluminosilicate (also known as cordierite), Ce-Zr mixed oxide, and Pd nanoparticles (Figure 5). Rare earth elements are well-known for their oxygen storage capability, which is mandatory for oxidation reaction of CO to CO2 [3].
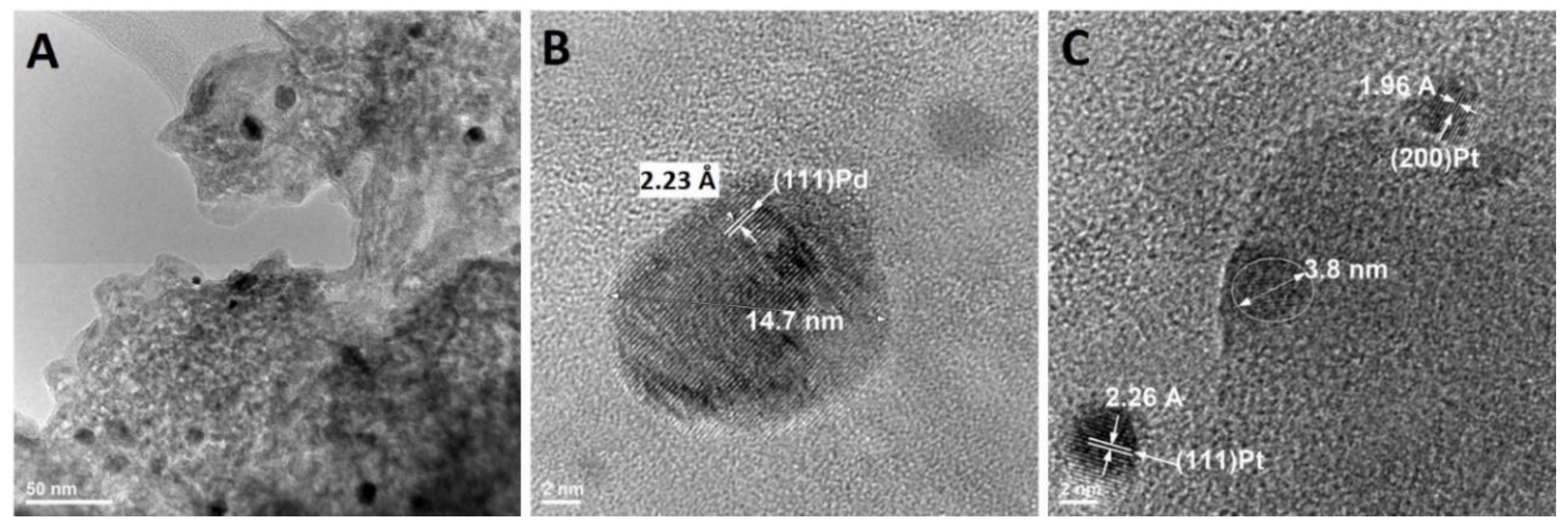
Figure 6 shows the morphology of the catalyst at the nanoscale. Dark-black spherical particles are supported on cordierite, present as a light grey material (Figure 6A). High resolution transmission electron microscopy (TEM) investigations show that there are two different types of nanoparticles (Figure 6B,C). Nanoparticles can be identified by their inter-planar distance, which is element specific. For example, the distance between two crystallographic planes (111) Pd is 2.23 Å, while for (111) Pt, it is 2.26 Å. Pd nanoparticles with diameters larger than 10 nm were observed throughout the investigation, while Pt nanoparticles were consistently found to be smaller than 5 nm. The absence of Pt in the XRD (Figure 5) can be explained based on the smaller size (i.e., 5 nm vs. 15 nm) and lower concentration (i.e., 0.014% vs. 0.046%). It is well-known that nanoparticle sintering is accelerated above their Tammann temperature, which can be approximated to half of the melting point on the absolute temperature scale [24]. Platinum’s Tammann temperature is 750 °C, which is 110 °C higher than Pd (640 °C). Catalytic convertors can reach 700 °C, which can explain the larger size of Pd nanoparticles. Indeed, the increase in Pd particle size is a well-known degradation concept in the automotive industry [3]. However, the electrochemical dissolution method outlined in Figure 7 and explained in detail in previous sections is designed to work for any platinum group metal of any size.
Electrodes were prepared by spraying a solution of spent automotive catalyst on Ti plates (Figure S4, Supporting Material). The potential step method was chosen owing to the fourfold improvement in the electrochemical dissolution (Figure 4B). All the electrochemical experiments were carried out at room temperature in 1 M HCl. The electrolyte composition was determined by ICP-OES at the beginning and at the end of 24 h dissolution experiments (Table 2). The Pt/Pd ratio in the pristine catalyst is approximately 0.3. The Pt/Pd ratio at the end of the dissolution experiment was determined to be 0.32, which is close to the initial value of 0.3. The dissolution efficiency can be determined by dividing the amount of Pt and/or Pd found in the electrolyte by the amount of Pt and/or Pd deposited on the electrode. The electrochemical dissolution efficiency for Pt and Pd was determined to be 47.1% and 48.7%, respectively (Table 2).
4. Discussion
Electrochemical methods were employed to investigate PGM electrochemical recycling. Initial experiments were carried out on polycrystalline Pt and the findings were transferred to a spent automotive catalyst that included Pd as well. The key findings in this work can be summarized as follows: (1) Pt dissolution is accelerated by the presence of chlorides and, to a lesser extent, by an acidic pH (Figure 2A); (2) the rate of Pt dissolution is constant for at least 7 continuous days (Figure 3A); (3) rapid changes in potential increase the dissolution by a factor of 4 (Figure 4B); and (4) the findings obtained on polycrystalline Pt can be extended to PGM recovery from spent automotive catalysts without any pretreatment.
There is a large body of work on the recovery of platinum group metals (PGMs) from spent catalytic structures that focus on achieving high recovery rates [5,23]. Different pretreatments of the catalysts are needed. Traditional methods that use HCl have a 25% and 35% leaching efficiency for Pt and Pd, respectively [8]. Some methods use elemental chlorine gas that reacts with the catalyst at 1000 °C to obtain recycling rates above 75% [25]. Other methods use HCl at high concentrations (i.e., 5 M), moderate temperatures (i.e., 80 °C), and pretreatment at 550 °C [26]. The use of chlorine at high temperatures should be performed with extreme precaution owing to its toxicity and extremely high reactivity. The United Nations Environment Programme (UNEP) recycling rates show that the overall PGM recovery rate is around 50% depending on the metal [27].
The effect of chloride ions on the electrochemical dissolution of Pt has been previously discussed [12,28]. The extensive dissolution of Pt in the presence of trace amounts of chloride is a well-known degradation mechanism in acidic polymer electrolyte membrane fuel cells [20]. The formation of chloro-complexes in HCl solutions has been exploited to recycle Pt from fuel cells [18,21,29]. The recovery of Pt from fuel cell electrodes is facilitated by the nature of the triple phase boundary, in which electrons, protons, and reactants meet at the Pt site. Moreover, the Pt loading on the carbon support is more than 50%. The backbone of automotive catalysts is the cordierite, which is non-conductive. However, the cordierite is covered by unreacted carbon that enables the electrochemical recycling of PGM.
Electrochemical techniques have been used to recover metals from different wastes with promising results [30,31]. There are very few reports [6,7] that make use of electrochemical tools to recover PGM from spent autocatalysts. The indirect production of chlorine gas by electro-generation seems to be a promising alternative to conventional pyro- and hydrometallurgy methods [7]. Chlorine evolution (R4) takes place at platinum electrodes. Figure 4B shows that chlorine gas alone is not sufficient to obtain high dissolution rates in our case. The subsequent oxidation and reduction of the PGM surface in chlorides is mandatory to obtain high dissolution rates. Moreover, Pt dissolution is accelerated by chlorides even at pH = 7 (Figure 2A), which can open the way to the electrochemical recycling of PGM in a non-corrosive environment, basically in brine. Further studies are needed to obtain high recovery rates at neutral pH.
5. Conclusions
The work at hand showed that platinum can be electrochemically recovered in different electrolytes. An acidic pH and the presence of chlorides are mandatory in achieving high dissolution rates. Platinum dissolution is achievable even at neutral pH, as long as the electrolyte contains chlorides. We made use of the knowledge in platinum dissolution to recycle the platinum group metals from spent automotive catalysts. Electrodes were rapidly manufactured by milling and spraying without any other pretreatment.
Electrochemical recycling of platinum group metals from spent catalytic structures was shown to have recovery rates as high as 50% in only 24 h. Increasing the amount of spent automotive catalysts to be treated is mandatory to forge an electrochemical recycling technology. Increasing the temperature is a simple way, although not desirable owing to issues of handling hot corrosive liquids. Other electrolytes can be used to further accelerate the dissolution. Designing electrochemical protocols that promote fast oxidation–reduction processes can significantly enhance dissolution rates.
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-4701/10/6/822/s1, Figure S1. Measured potentials of Hg/Hg2Cl2, Ag/AgCl, and Hg/Hg2SO4; Figure S2. Calculation of electrochemical surface area for the peaks of CVs in 0.5M K2SO4; Figure S3. UV/vis spectrum of the solution from 7 days of experiment; Figure S4. Preparation of solution with catalytic converter steps; Table S1. The potentials of reference electrodes; Table S2. Operating parameters for ICP-OES; Table S3. Percent interference for matrix elements of SRM 2557 in the standard resolution ICP-OES. Individual standards for each element were analyzed at Pt wavelengths at expected concentrations found in NIST SRM 2557, assuming a sample loading of 2 g into 50 mL. The ratio of interference to analyte is recorded as a percentage for each potential analyte; Table S4. The roughness factor for electrolytes.
Author Contributions
C.D. and S.N.S., concept and methodology; C.D. and F.I.M., electrochemical experiments; C.D. analyzed the results; A.C. and E.V. characterized the catalyst; C.D. wrote the first draft; E.V and S.N.S. reviewed and edited the draft; R.T., visualization; supervision was done by V.F., E.V., and S.N.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant of the Romanian Ministery of Research and Innovation, CCCDI UEFISCDI, project number PCCDI 76/2018 and PD 111/2018 within PNCDI III. This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreements No. 797781 (CALSOL). The results of this publication reflect only the authors’ view and the Commission is not responsible for any use that may be made of the information it contains.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eu-law-Eur-lex. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:52017DC0490&from=EN (accessed on 10 May 2020).
- Mudd, G.M.; Jowitt, S.M.; Werner, T.T. Global platinum group element resources, reserves and mining—A critical assessment. Sci. Total Environ. 2018, 622, 614–625. [Google Scholar] [CrossRef]
- Shelef, M.; Mccabe, R.W. Twenty-five years after introduction of automotive catalysts: What next? Catal. Today 2000, 62, 35–50. [Google Scholar] [CrossRef]
- Hagelüken, C. Recycling of (critical) metals. In Critical Metals Handbook; Gunn, G., Ed.; John Wiley & Sons: Chichester, UK, 2013; pp. 41–69. [Google Scholar]
- Dong, H.; Zhao, J.; Chen, J.; Wu, Y.; Li, B. Recovery of platinum group metals from spent catalysts: A review. Int. J. Miner. Process. 2015, 145, 108–113. [Google Scholar] [CrossRef]
- Saguru, C.; Ndlovu, S.; Moropeng, D. A review of recent studies into hydrometallurgical methods for recovering PGMs from used catalytic converters. Hydrometallurgy 2018, 182, 44–56. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, J.C.; Park, S.W.; Jeong, J.; Kumar, V. Dissolution behaviour of platinum by electro-generated chlorine in hydrochloric acid solution. J. Chem. Technol. Biotechnol. 2013, 88, 1212–1219. [Google Scholar] [CrossRef]
- Wei, X.; Liu, C.; Cao, H.; Ning, P.; Jin, W.; Yang, Z.; Wang, H.; Sun, Z. Understanding the features of PGMs in spent ternary automobile catalysts for development of cleaner recovery technology. J. Clean. Prod. 2019, 239, 118031. [Google Scholar] [CrossRef]
- Fornalczyk, A.; Saternus, M. Platinum Recovery From Used Auto Catalytic. Metalurgia 2013, 52, 219–222. [Google Scholar]
- Carmo, M.; Stolten, D. Energy Storage Using Hydrogen Produced From Excess Renewable Electricity: Power to Hydrogen. In Science and Engineering of Hydrogen-Based Energy Technologies; Elsevier Inc.: London, UK, 2019; pp. 165–199. [Google Scholar] [CrossRef]
- Wang, S.; Tarroja, B.; Smith, L.; Sha, B.; Samuelsen, S. Prioritizing among the end uses of excess renewable energy for cost-effective greenhouse gas emission reductions. Appl. Energy 2019, 235, 284–298. [Google Scholar] [CrossRef]
- Geiger, S.; Cherevko, S.; Mayrhofer, K.J.J. Dissolution of Platinum in Presence of Chloride Traces. Electrochim. Acta 2015, 179, 24–31. [Google Scholar] [CrossRef]
- Bard, A.J. Standard Potentials in Aqueous Solution; IUPAC: Oxford, UK, 1985; pp. 353–354. [Google Scholar]
- Jerkiewicz, G.; Vatankhah, G.; Lessard, J.; Soriaga, M.P.; Park, Y.S. Surface-oxide growth at platinum electrodes in aqueous H2SO4: Reexamination of its mechanism through combined cyclic-voltammetry, electrochemical quartz-crystal nanobalance, and Auger electron spectroscopy measurements. Electrochim. Acta 2003, 49, 1451–1459. [Google Scholar] [CrossRef]
- Kinoshita, K.; Lundquist, J.; Stonehart, P. Hydrogen adsorption on high surface area platinum crystallites. J. Catal. 1973, 31, 325–334. [Google Scholar] [CrossRef]
- Mundy, G.R.; Potter, R.J.; Christensen, P.A.; Hamnett, A. A study of the electro-oxidation of methanol on platinum in 1 M Na2SO4 by electrochemical and in-situ FTIR techniques. J. Electroanal. Chem. 1990, 279, 257–272. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-esparza, A.T.; Takanabe, K. Mechanistic Switching by Hydronium Ion Activity for Hydrogen Evolution and Oxidation over Polycrystalline Platinum Disk and Platinum/Carbon Electrodes. ChemElectroChem 2014, 1, 1497–1507. [Google Scholar] [CrossRef]
- Nørgaard, C.F.; Stamatin, S.N.; Skou, E.M. Redeposition of electrochemically dissolved platinum as nanoparticles on carbon. Int. J. Hydrog. Energy 2014, 39, 17322–17326. [Google Scholar] [CrossRef]
- Sharma, H.; Sharma, V.; Huan, T.D. Exploring PtSO4 and PdSO4 phases: An evolutionary algorithm based investigation. Phys. Chem. Chem. Phys. 2015, 17, 18146–18151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalov, A.A.; Katsounaros, I.; Auinger, M.; Cherevko, S.; Meier, J.C.; Klemm, S.O.; Mayrhofer, K.J.J. Dissolution of platinum: Limits for the deployment of electrochemical energy conversion? Angew. Chem. Int. Ed. Engl. 2012, 51, 12613–12615. [Google Scholar] [CrossRef] [Green Version]
- Hodnik, N.; Baldizzone, C.; Polymeros, G.; Geiger, S.; Grote, J.; Cherevko, S.; Mingers, A.; Zeradjanin, A.; Mayrhofer, K.J.J. Platinum recycling going green via induced surface potential alteration enabling fast and efficient dissolution. Nat. Commun. 2016, 7, 1–6. [Google Scholar] [CrossRef]
- He, J.-J.; Wang, C.-X.; Zheng, T.-T.; Zhao, Y.-K. Thermally Induced Deactivation and the Corresponding Strategies for Improving Durability in Automotive Three-Way Catalysts: A review of latest developments and fundamentals. In Johnson Matthey Technology Review; Johnson Matthey Plc: Royston, UK, 2016; pp. 179–185. [Google Scholar] [CrossRef]
- Jha, M.K.; Lee, J.C.; Kim, M.S.; Jeong, J.; Kim, B.S.; Kumar, V. Hydrometallurgical recovery/recycling of platinum by the leaching of spent catalysts: A review. Hydrometallurgy 2013, 133, 23–32. [Google Scholar] [CrossRef]
- Satterfield, C.N. Heterogeneous Catalysis in Industrial Practice, 2nd ed.; Krieger Publishing: Melbourne, FL, USA, 1996. [Google Scholar]
- Bronshtein, I.; Feldman, Y.; Shilstein, S.; Wachtel, E.; Lubomirsky, I.; Kaplan, V. Efficient Chloride Salt Extraction of Platinum Group Metals from Spent Catalysts. J. Sustain. Metall. 2018, 4, 103–114. [Google Scholar] [CrossRef]
- Prasetyo, E.; Anderson, C. Platinum Group Elements Recovery from Used Catalytic Converters by Acidic Fusion and Leaching. Metals 2020, 10, 485. [Google Scholar] [CrossRef] [Green Version]
- Graedel, T.E.; Allwood, J.; Birat, J.-P.; Reck, B.K.; Sibley, S.F.; Sonnemann, G.; Buchert, M.; Hagelüken, C. UNEP Recycling Rates of Metals–A Status Report, A Report of the Working Group on the Global Metal Flows to the International Resource Panel. 2011. Available online: https://www.resourcepanel.org/reports/recycling-rates-metals (accessed on 12 April 2020).
- Shrestha, B.R.; Tada, E.; Nishikata, A. Effect of chloride on platinum dissolution. Electrochim. Acta 2014, 143, 161–167. [Google Scholar] [CrossRef]
- Sharma, R.; Gyergyek, S.; Andersen, S.M. Environmentally and Industrially Friendly Recycling of Platinum Nanoparticles Through Electrochemical Dissolution–Electrodeposition in Acid-Free/Dilute Acidic Electrolytes. ChemSusChem 2018, 11, 3742–3750. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, N.; Legeai, S.; Balva, M.; Hazotte, C.; Comel, J.; Lapicque, F.; Billy, E.; Meux, E. Recovery of Metals from Secondary Raw Materials by Coupled Electroleaching and Electrodeposition in Aqueous or Ionic Liquid Media. Metals 2018, 8, 556. [Google Scholar] [CrossRef] [Green Version]
- Balva, M.; Legeai, S.; Leclerc, N.; Billy, E.; Meux, E. Environmentally Friendly Recycling of Fuel-Cell Membrane Electrode Assemblies by Using Ionic Liquids. ChemSusChem 2014, 10, 2922–2935. [Google Scholar] [CrossRef]
Figure 1.
Cyclic voltammograms (CVs) recorded with polycrystalline platinum disk electrode with a surface area of 0.785 cm2 at 100 mV/s in acidic and neutral media for 24 h of the dissolution study: (A) 1 M HCl, reference electrode (RE): Hg/Hg2Cl2; (B) 0.5 M H2SO4, RE: Hg/Hg2SO4; (C) 1 M KCl, RE: Hg/Hg2Cl2; (D) 0.5 M K2SO4, RE: Hg/Hg2SO4.
Figure 1.
Cyclic voltammograms (CVs) recorded with polycrystalline platinum disk electrode with a surface area of 0.785 cm2 at 100 mV/s in acidic and neutral media for 24 h of the dissolution study: (A) 1 M HCl, reference electrode (RE): Hg/Hg2Cl2; (B) 0.5 M H2SO4, RE: Hg/Hg2SO4; (C) 1 M KCl, RE: Hg/Hg2Cl2; (D) 0.5 M K2SO4, RE: Hg/Hg2SO4.

Figure 2.
(A) Amount of dissolved Pt during 24 h for each electrolyte solution; (B) UV/vis absorbance spectra of the electrolytes recorded after the dissolution of the platinum disk electrode.
Figure 2.
(A) Amount of dissolved Pt during 24 h for each electrolyte solution; (B) UV/vis absorbance spectra of the electrolytes recorded after the dissolution of the platinum disk electrode.

Figure 3.
(A) Amount of platinum dissolved for each day determined by ICP-OES; (B) absorption intensity at 262 nm (red circles), integrated area under the peak (black squares), and the amount of dissolved Pt determine by ICP-OES (blue triangles).
Figure 3.
(A) Amount of platinum dissolved for each day determined by ICP-OES; (B) absorption intensity at 262 nm (red circles), integrated area under the peak (black squares), and the amount of dissolved Pt determine by ICP-OES (blue triangles).

Figure 4.
(A) Pt dissolved electrochemically in a rotating disc electrode (0.0314 cm−2) setup in air; (B) the amount of Pt dissolution by cyclic voltammetry (CV), chronoamperometry-potential step (PS) with a 3 s step at 0.45 and 1.25 V vs. RHE (PS), potential hold (PH) at 0.45 V vs. RHE for 24 h, and PH at 1.25 V vs. RHE. Electrolyte: 1M HCl, RE: Ag/AgCl.
Figure 4.
(A) Pt dissolved electrochemically in a rotating disc electrode (0.0314 cm−2) setup in air; (B) the amount of Pt dissolution by cyclic voltammetry (CV), chronoamperometry-potential step (PS) with a 3 s step at 0.45 and 1.25 V vs. RHE (PS), potential hold (PH) at 0.45 V vs. RHE for 24 h, and PH at 1.25 V vs. RHE. Electrolyte: 1M HCl, RE: Ag/AgCl.
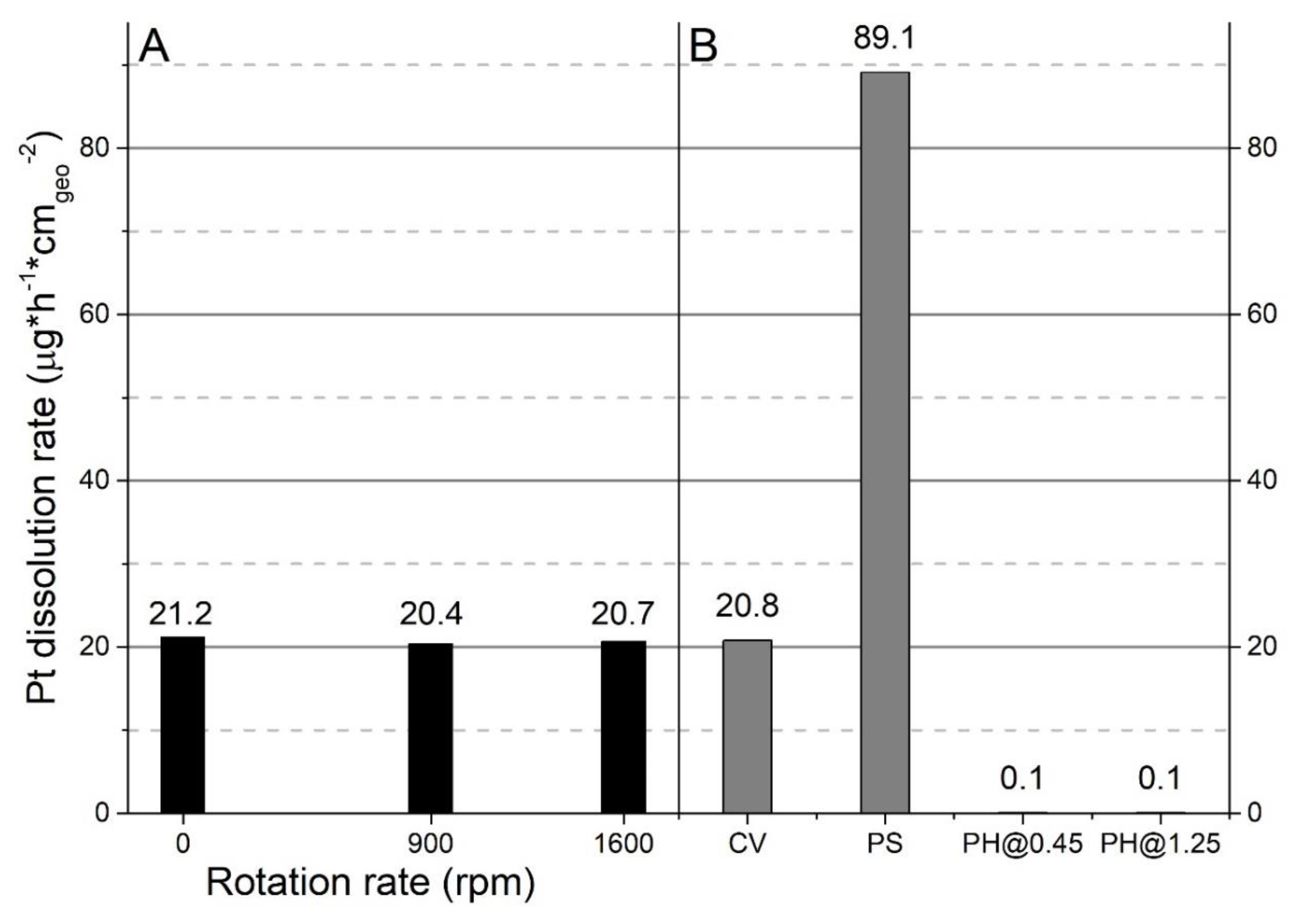
Figure 5.
Powder X-ray diffraction of spent automotive catalyst.
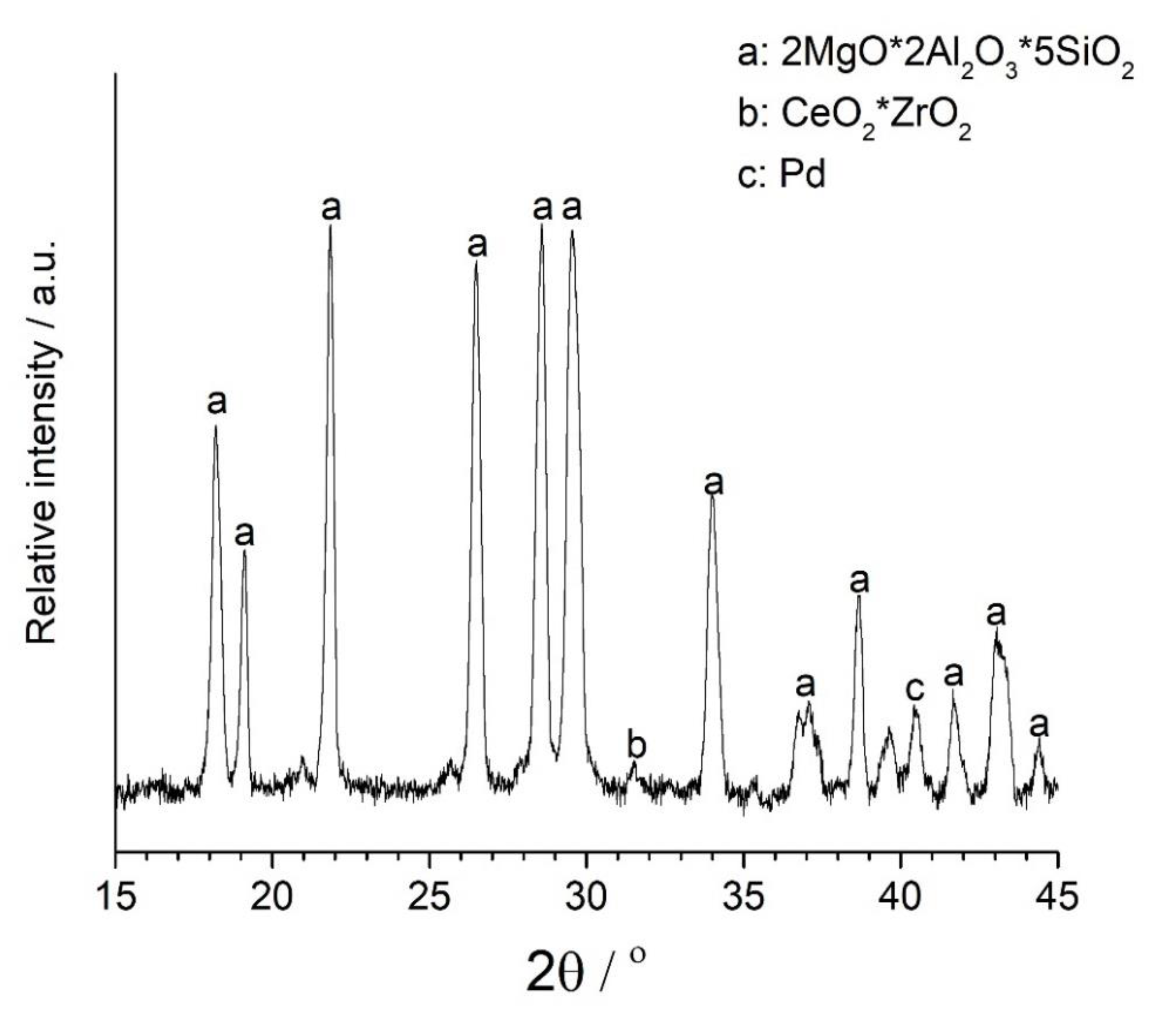
Figure 6.
High resolution transmission electron micrographs of the spent automotive catalyst (A), Pd nanoparticles (B), and Pt nanoparticles (C) with their associated inter planar distance.
Figure 6.
High resolution transmission electron micrographs of the spent automotive catalyst (A), Pd nanoparticles (B), and Pt nanoparticles (C) with their associated inter planar distance.
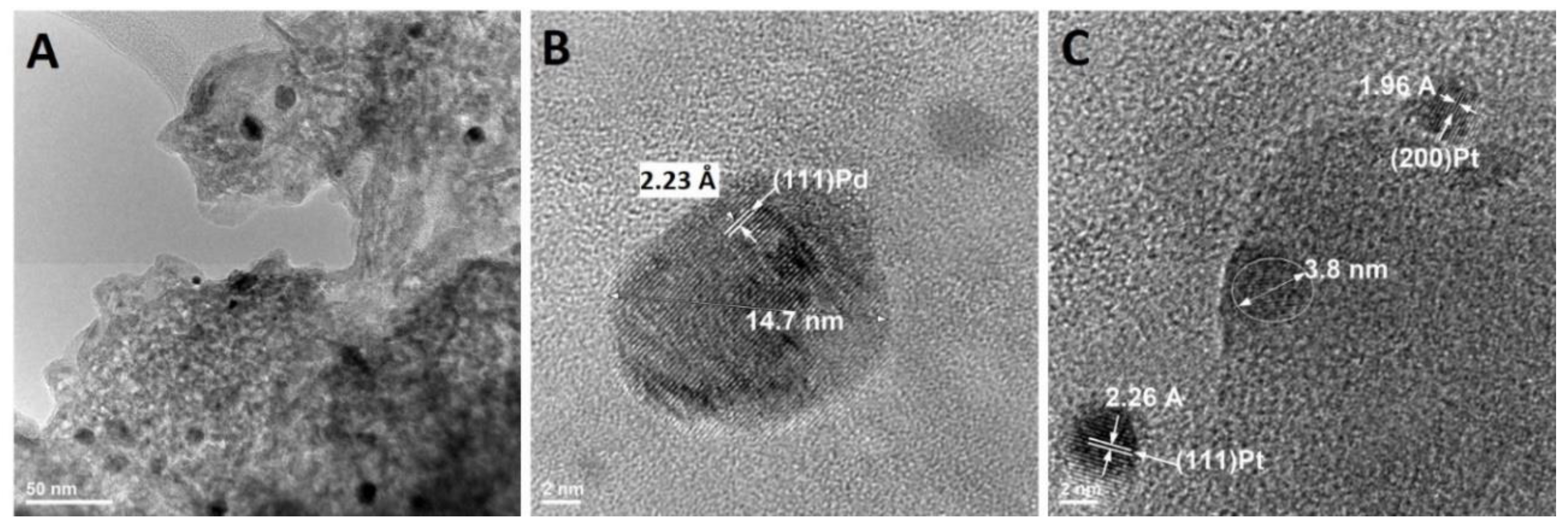
Figure 7.
The process of electrochemical dissolution of platinum.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Electrochemical potential of different reactions, data from [13].
Table 1.
Electrochemical potential of different reactions, data from [13].
| Reactions | Potential (V) vs. Standard Hydrogen Electrode (SHE) | Abbreviations |
|---|---|---|
| 1.188 | R1 | |
| ) | 0.758 | R2 |
| ) | 0.744 | R3 |
| 1.396 | R4 | |
| 1.229 | R5 | |
| 0.980 | R6 |
Table 2.
Electrochemical dissolution of spent automotive catalyst in 1 M HCl with chronoamperometry-potential step (PS) experiments with a 3 s step at 0.6 V and 1.5 V vs. RHE.
Table 2.
Electrochemical dissolution of spent automotive catalyst in 1 M HCl with chronoamperometry-potential step (PS) experiments with a 3 s step at 0.6 V and 1.5 V vs. RHE.
| Catalyst/μg | Initially/μg | At the End of Experiment/μg | Element Efficiency/% | Total Efficiency/% | |||
|---|---|---|---|---|---|---|---|
| Pt | Pd | Pt | Pd | Pt | Pd | Pt and Pd | |
| 7075 | 1.061 | 3.184 | 0.5 | 1.55 | 47.1 | 48.7 | 48.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Diac, C.; Maxim, F.I.; Tirca, R.; Ciocanea, A.; Filip, V.; Vasile, E.; Stamatin, S.N. Electrochemical Recycling of Platinum Group Metals from Spent Catalytic Converters. Metals 2020, 10, 822. https://doi.org/10.3390/met10060822
AMA Style
Diac C, Maxim FI, Tirca R, Ciocanea A, Filip V, Vasile E, Stamatin SN. Electrochemical Recycling of Platinum Group Metals from Spent Catalytic Converters. Metals. 2020; 10(6):822. https://doi.org/10.3390/met10060822
Chicago/Turabian StyleDiac, Cornelia, Florentina Iuliana Maxim, Radu Tirca, Adrian Ciocanea, Valeriu Filip, Eugeniu Vasile, and Serban N. Stamatin. 2020. "Electrochemical Recycling of Platinum Group Metals from Spent Catalytic Converters" Metals 10, no. 6: 822. https://doi.org/10.3390/met10060822
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.