
Electrochemical Investigation of Lateritic Ore Leaching Solutions for Ni and Co Ions Extraction

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
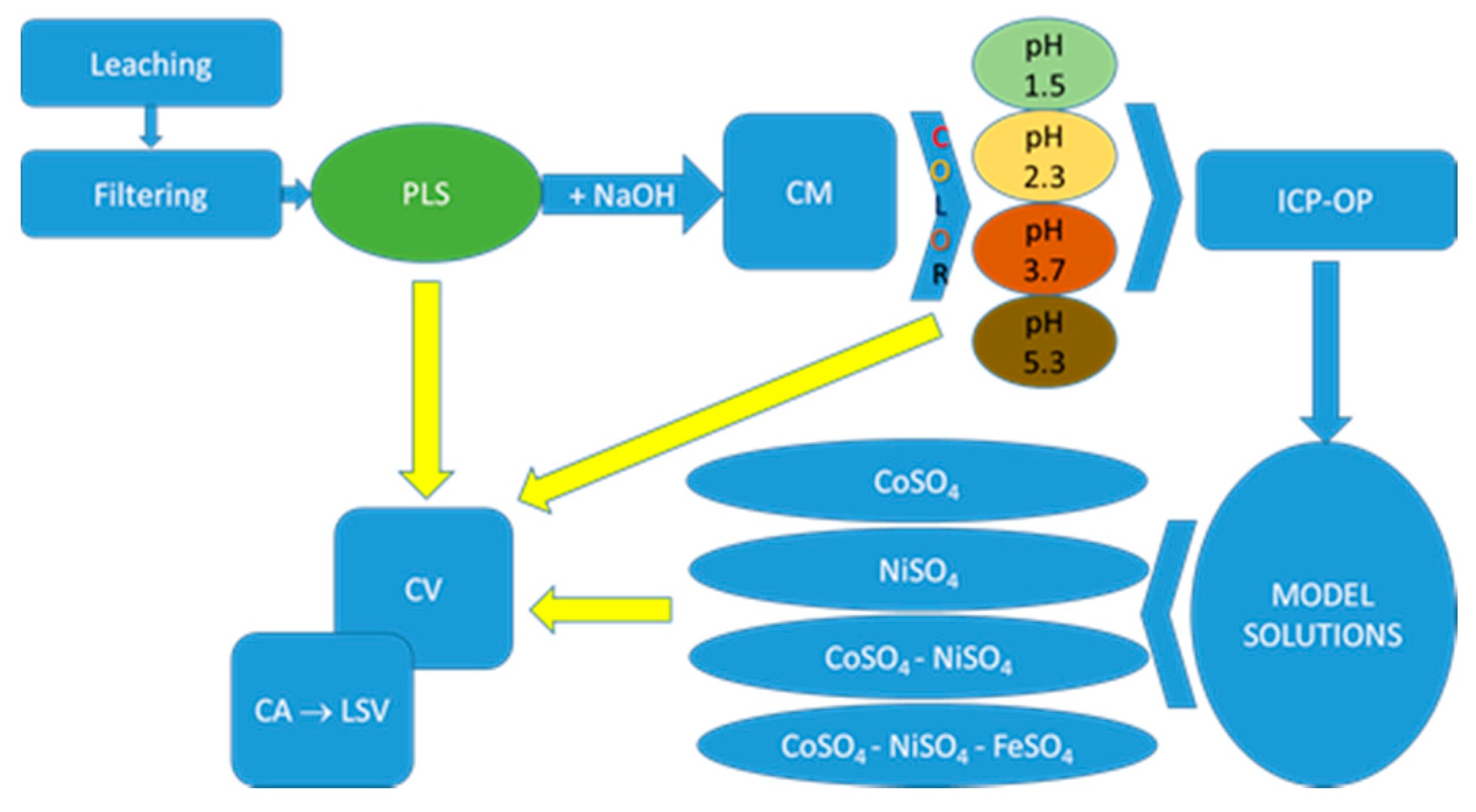
2.1. Leaching
2.2. Preparation of Model Solutions
2.3. Electrochemical Methods
- Glassy carbon electrode (GC) was the working electrode for cyclic voltammetry (CV) measurements in PLSs, CoSO4 and NiSO4 model solutions, as well as in base solution. The working electrode was mechanically polished consecutively on emery papers of the following grades: 280, 360, 800 and 1000, and then on polishing cloths (Buehler Ltd., Lake Bluff, IL, USA) impregnated with alumina of 1, 0.3 and 0.05 µm grades;
- For the examination of the electrochemical potentiostatic extracts from PLSs at different pH, working electrodes were the alloys deposited onto polished GC;
- To model the electrochemical dissolution by LSV, working electrodes were Ni–, Co–, Fe–, Ni–Co and Ni–Co–Fe alloys deposited on GC from the model solutions. Before each electrodeposition, the GC surface was mechanically polished with a polishing cloth (Buehler Ltd., Lake Bluff, IL, USA) impregnated with a water suspension of alumina powder (0.05 μm grade). Characterization of alloy composition was performed by alloy electrochemical dynamic dissolution using a linear sweep voltammetry (LSV) technique.
3. Results and Discussion
3.1. Conductometric Measurements
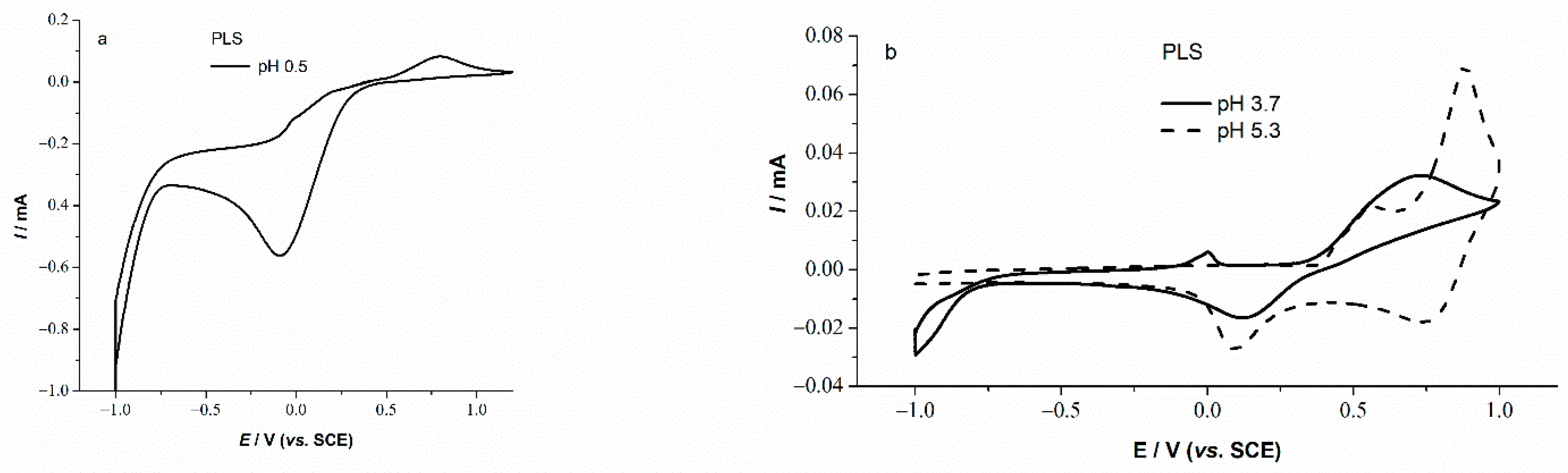
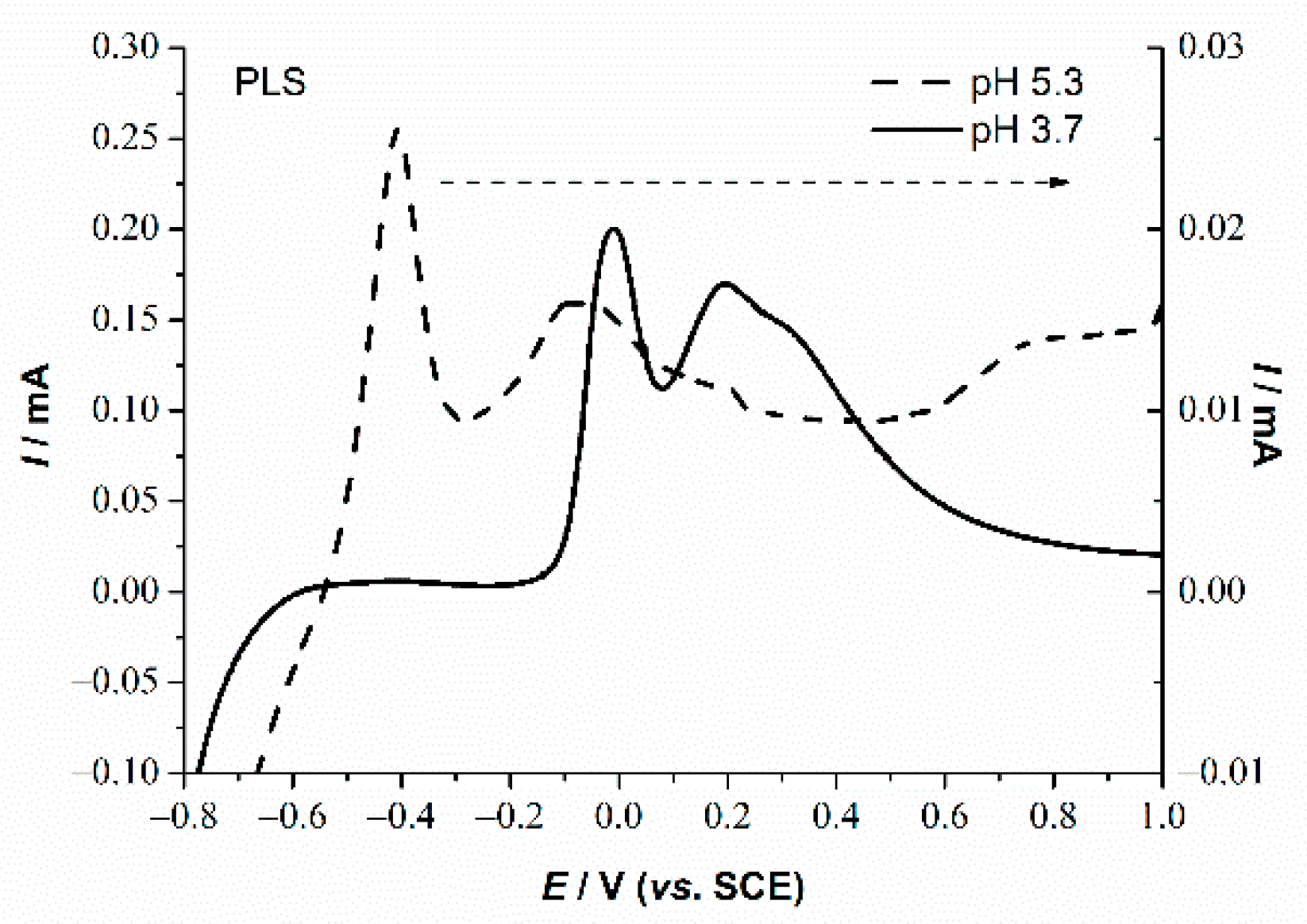
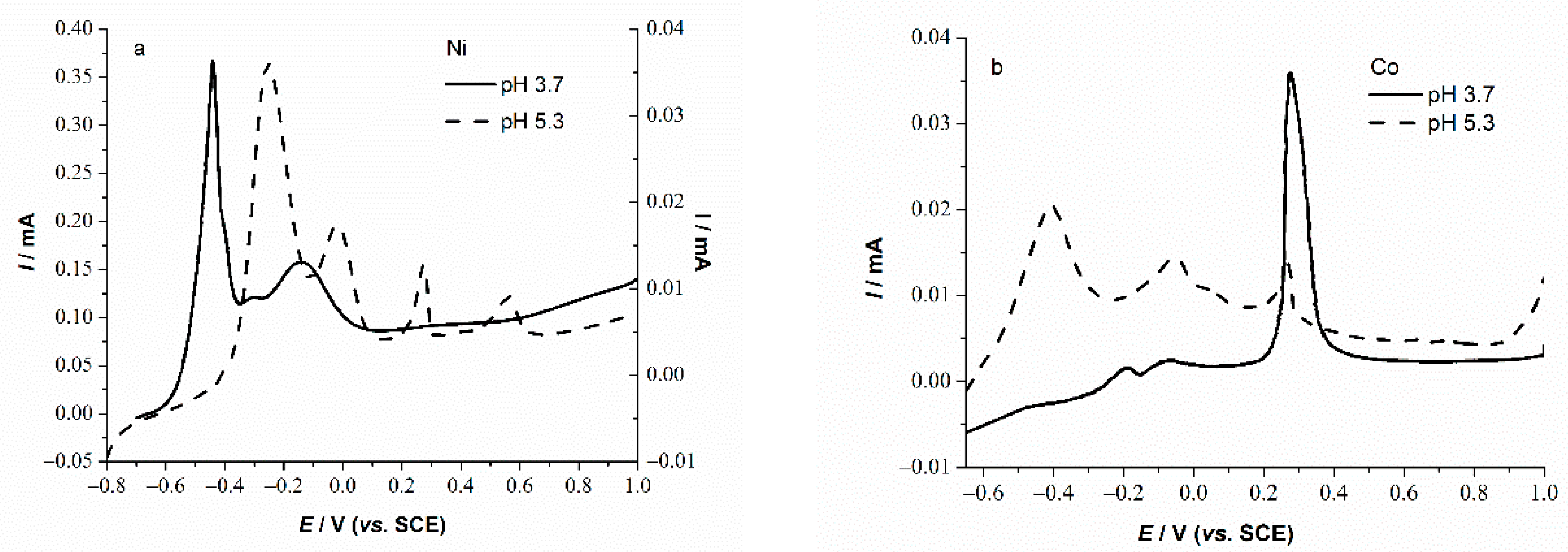
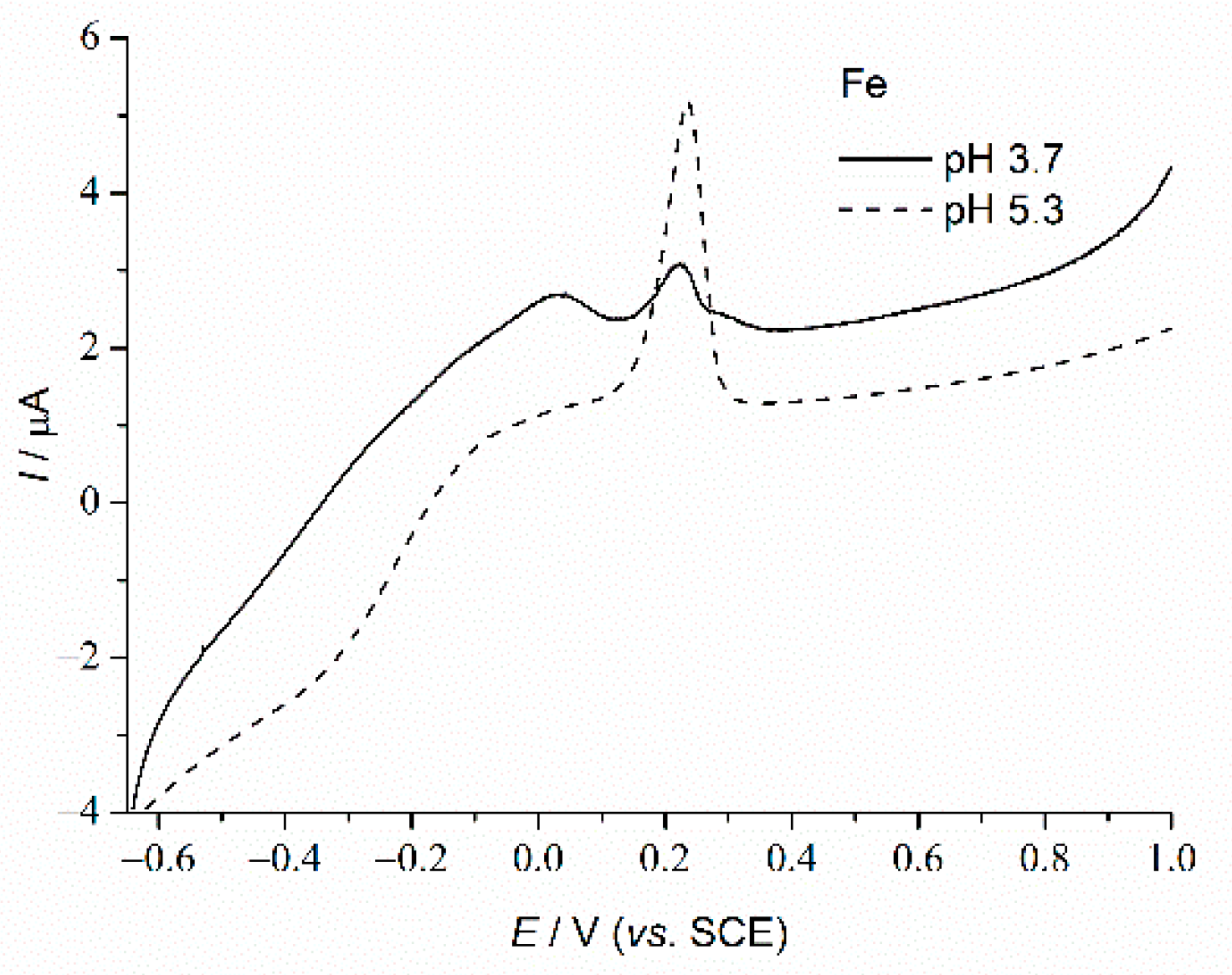
3.2. Basic Electrochemical Behavior of PLS
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slack, J.F.; Kimball, B.E.; Shedd, K.B. Cobalt. In Critical Mineral Resources of the United States—Economic and Environ Mental Geology and Prospects for Future Supply; Schulz, K.J., DeYoung, J.H., Seal, R.R., Bradley, D.C., Eds.; USGS Pubs: Reston, VA, USA, 2017; Volume 1082, pp. F1–F40. [Google Scholar]
- Bloomberg. Available online: https://www.bloomberg.com/news/articles/2021-02-25/musk-says-nickel-is-biggest-concern-for-electric-car-batteries (accessed on 12 December 2021).
- Wood, M.; Li, J.; Ruthera, R.E.; Du, Z.; Self, E.C.; Meyer, H.M., III; Daniel, C.; Belharouak, I.; Wood, D.L., III. Chemical stability and long-term cell performance of low-cobalt, Ni-Rich cathodes prepared by aqueous processing for high-energy Li-Ion batteries. Energy Stor. Mater. 2020, 24, 188–197. [Google Scholar] [CrossRef]
- Deetman, S.; Pauliuk, S.; van Vuuren, D.P.; van der Voet, E.; Tukker, A. Scenarios for demand growth of metals in electricity generation technologies, cars, and electronic appliances. Environ. Sci. Technol. 2018, 52, 4950–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.R. (Ed.) Nickel, Cobalt, and Their Alloys, ASM Specialty Handbook; ASM International: Novelty, OH, USA, 2000. [Google Scholar]
- Statista. Available online: https://www.statista.com/statistics/967700/global-demand-for-nickel-in-ev-batteries/ (accessed on 12 December 2021).
- Banza, A.N.; Gock, E.; Kongolo, K. Base metals recovery from copper smelter slag by oxidising leaching and solvent extraction. Hydrometallurgy 2002, 67, 63–69. [Google Scholar] [CrossRef]
- Altundogan, H.S.; Boyrazli, M.; Tumen, F. A study on the sulphuric acid leaching of copper converter slag in the presence of dichromate. Miner. Eng. 2004, 17, 465–467. [Google Scholar] [CrossRef]
- Song, S.; Sun, W.; Wang, L.; Liu, R.; Han, H.; Hu, Y.; Yang, Y. Recovery of cobalt and zinc from the leaching solution of zinc smelting slag. J. Environ. Chem. Eng. 2019, 7, 102777. [Google Scholar]
- Khalid, M.K.; Hamuyuni, J.; Agarwal, V.; Pihlasalo, J.; Haapalainen, M.; Lundström, M. Sulfuric acid leaching for capturing value from copper rich converter slag. J. Clean. Prod. 2019, 215, 1005–1013. [Google Scholar] [CrossRef]
- Meshram, P.; Prakash, U.; Bhagat, L.; Abhilash; Zhao, H.; van Hullebucsh, E.D. Processing of Waste Copper Converter Slag Using Organic Acids for Extraction of Copper, Nickel, and Cobalt. Minerals 2020, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Potysz, A.; Lens, P.N.; van de Vossenberg, J.; Rene, E.R.; Grybos, M.; Guibaud, G.; van Hullebusch, E.D. Comparison of Cu, Zn and Fe bioleaching from Cu-metallurgical slags in the presence of Pseudomonas fluorescens and Acidithiobacillus thiooxidans. Appl. Geochem. 2016, 68, 39–52. [Google Scholar] [CrossRef]
- Yang, Z.; Rui-lin, M.; Wang-dong, N.; Hui, W. Selective leaching of base metals from copper smelter slag. Hydrometallurgy 2010, 103, 25–29. [Google Scholar] [CrossRef]
- Elias, M. Nickel laterite deposit—Geological overview, resources and exploitation. In Giant Ore Deposits: Characteristics, Genesis and Exploration; Cooke, D., Pongratz, J., Eds.; Special Publication 4; University of Tasmania, Centre for Ore Deposit Research: Hobart, Australia, 2002; Volume 6, pp. 205–220. [Google Scholar]
- Koza, J.M.; Uhlemann, M.; Gebert, A.; Schultz, L. The effect of a magnetic field on the pH value in front of the electrode surface during the electrodeposition of Co, Fe and CoFe alloys. J. Electroanal. Chem. 2008, 617, 194–202. [Google Scholar] [CrossRef]
- Macdonald, D.D.; Liang, R.Y.; Pound, B.G. An Electrochemical Impedance Study of the Passive Film on Single Crystal Ni(111)in Phosphate Solutions. J. Electrochem. Soc. 1987, 134, 2981. [Google Scholar] [CrossRef]
- Martini, E.M.A.; Amaral, S.T.; Muller, I.L. Electrochemical behaviour of Invar in phosphate solutions at pH 6.0. Corros. Sci. 2004, 46, 2097–2115. [Google Scholar] [CrossRef]
- Chivot, J.; Mendoza, L.; Mansour, C.; Pauporte´, T.; Cassir, M. New insight in the behaviour of Co–H2O system at 25–150 °C, based on revised Pourbaix diagrams. Corros. Sci. 2008, 50, 62–69. [Google Scholar] [CrossRef]
- Yu, Y.; Sun, L.; Ge, H.; Wei, G.; Jiang, L. Study on Electrochemistry and Nucleation Process of Nickel Electrodeposition. Int. J. Electrochem. Sci. 2017, 12, 485–495. [Google Scholar] [CrossRef]
- Tian, L.; Xu, J.; Qiang, C. The electrodeposition behaviors and magnetic properties of Ni–Co films. Appl. Surf. Sci. 2011, 257, 4689–4694. [Google Scholar] [CrossRef]
- Yu, Y.; Song, Z.; Ge, H.; Wei, G.; Jian, L. Electrochemical mechanism of cobalt film electrodeposition process. Mater. Res. Innov. 2016, 20, 280–284. [Google Scholar] [CrossRef]
- Richardson, J.A.; Abdullahi, A.A. Corrosion in Alkalis, Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SiO2 | MgO | Fe2O3 | NiO | Al2O3 | CaO | MnO | Cr2O3 | CuO | Co3O4 | K2O | TiO2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 11.70 | 5.81 | 70.70 | 3.80 | 4.27 | 0.13 | 0.73 | 1.78 | <DL 1 | 0.05 | 0.02 | 0.07 |
| pH | Co | Ni | Fe |
|---|---|---|---|
| 3.7 | 0.3424 | 13.5 | 4.8 |
| 5.3 | 0.2661 | 12.3 | 0.0464 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitterecker, J.; Košević, M.; Stopic, S.; Friedrich, B.; Panić, V.; Stevanović, J.; Mihailović, M. Electrochemical Investigation of Lateritic Ore Leaching Solutions for Ni and Co Ions Extraction. Metals 2022, 12, 325. https://doi.org/10.3390/met12020325
Mitterecker J, Košević M, Stopic S, Friedrich B, Panić V, Stevanović J, Mihailović M. Electrochemical Investigation of Lateritic Ore Leaching Solutions for Ni and Co Ions Extraction. Metals. 2022; 12(2):325. https://doi.org/10.3390/met12020325
Chicago/Turabian StyleMitterecker, Jonas, Milica Košević, Srecko Stopic, Bernd Friedrich, Vladimir Panić, Jasmina Stevanović, and Marija Mihailović. 2022. "Electrochemical Investigation of Lateritic Ore Leaching Solutions for Ni and Co Ions Extraction" Metals 12, no. 2: 325. https://doi.org/10.3390/met12020325