3.1. Structural Properties
HfX (X = Os, Ir and Pt) compounds are in the (CsCl type) Pm3m space group, correspondingly. In an HfX unit cell, an Hf atom locates at 1a (0, 0, 0) and an X atom stays at 1b (0.5, 0.5, 0.5). In order to obtain structural properties of HfX compounds, the geometric optimizations are made firstly. The equilibrium lattice constants using both GGA and LDA methods are tabulated in
Table 1, along with the available experimental [
10,
11,
12] and theoretical [
6,
13,
14] values for reference. Generally, the qualities for structural optimizations in HfX compounds using both GGA and LDA methods are comparable and acceptable. In detail, the optimized lattice constants for HfOs and HfIr are in good agreement with theoretical values [
6,
13,
14], respectively. For HfOs and HfIr, the LDA computed values are more approaching to the experimental value. On the other aspect, the GGA method can give better optimized values than the LDA method in HfPt. Therefore, either method shows obvious superiority in the structural optimization of HfX compounds. Resultantly, both GGA and LDA methods are used in the following theoretical studies for HfX compounds.
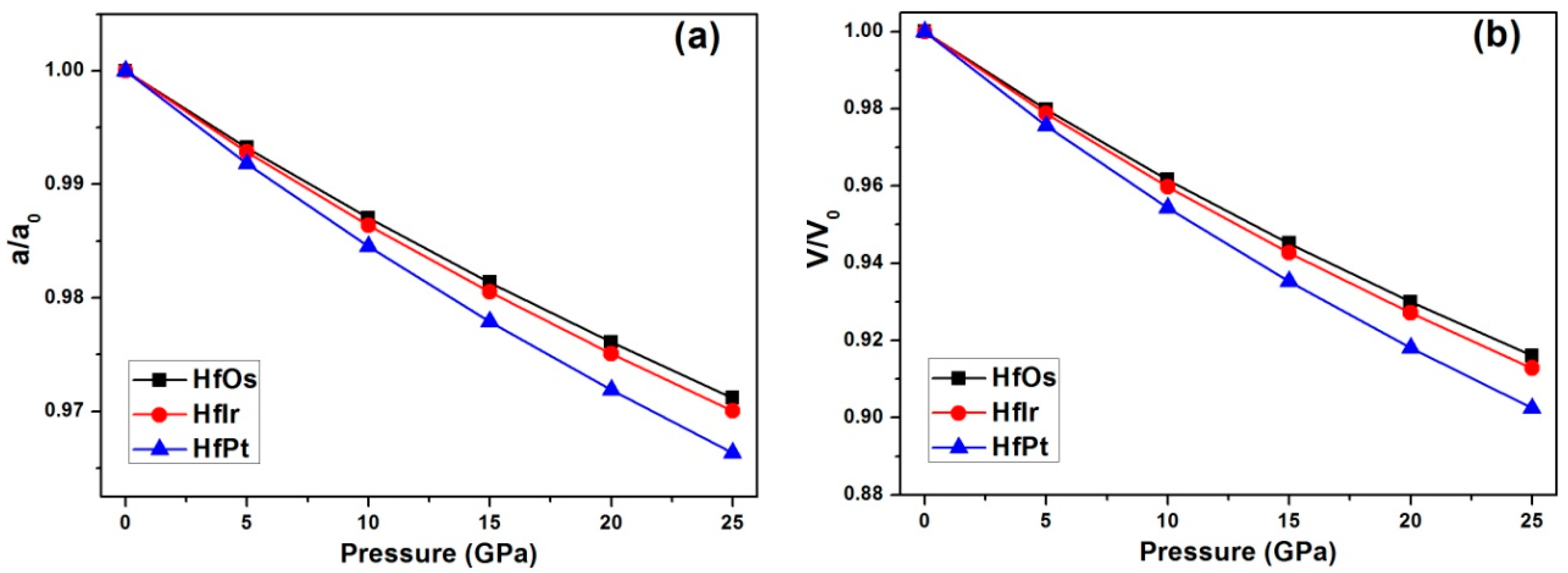
Under increasing pressures from 0 to 25 GPa with per step of 5 GPa, the relative changes of (a) lattice constant and (b) unit cell volume for HfX compounds using GGA method are exhibited in
Figure 1. In
Figure 1a, the relative change of lattice constant (
a) for HfPt is alternated larger than HfIr and HfOs under growing pressures. Similarly, the unit cell volume (V) changes larger in HfPt than HfIr and HfOs under pressures (
Figure 1b). Therefore, the obtained pressure volume curve for each compound is formulated to a third-order Birch-Murnaghan equation of state (EOS) [
26]:
For HfOs, HfIr and HfPt, the fitted bulk moduli (
B0) are 234.3 GPa, 220.9 GPa and 191.9 GPa, and their pressure derivatives (
B0’) are 4.43, 4.66 and 4.64, accordingly. As a result, the bulk moduli are in the sequence of HfOs > HfIr > HfPt. Using LDA method, the bulk moduli are still in the order of HfOs > HfIr > HfPt, as exhibited in
Table 1.
The thermodynamic stability is closely associated with formation enthalpy in binary compounds. To evaluate the thermodynamic stability of a compound, the formation enthalpy (
Hf) is expressed by the following equation [
27,
28]:
where
is the total energy of an HfX unit cell including an Hf atom and an X atom with equilibrium lattice parameters (X = Os, Ir and Pt);
EHf and
EX are the total energy per atom of pure element solids at their ground states. Hf and Os are HCP metals, and Ir and Pt are FCC metals at ground state.
The negative formation enthalpy has denoted that the chemical process is exothermic, indicating the stability of resulted compounds. Moreover, the larger negative formation enthalpy has signified better stability of a compound [
29]. In
Table 1, the formation enthalpies of HfX compounds are tabulated. Generally, HfX compounds should be stable owing to their negative formation enthalpies derived from GGA and LDA methods. Furthermore, their thermodynamic stabilities are both in the order of HfPt > HfIr > HfOs. This conclusion is the same with Xing’s work [
6]. In detail, the
Hf values calculated by GGA method are typically more negative than LDA method. For HfOs, the calculated H
f value by GGA method is in good consistency with experimental value [
24] and theoretical value from Liu’s work [
14], but smaller than Xing’s [
6] and Levy’s [
3] reports. For HfIr and HfPt, our calculated
Hf values are both smaller than theoretical values from Xing’s work [
6] and experimental values from Gachon’s report [
25]. Nevertheless, the crystal structures for HfIr and HfPt are not mentioned in this experimental work [
25], which has degraded the reference value of this experimental work.
3.3. Elastic Properties
The elastic constants and elastic moduli are the critical information to study the mechanical properties of compounds. A full set of elastic constant for cubic crystal, including
C11,
C12 and
C44, can be achieved using the stress-train method [
36] (
Table 2). Observably, the elastic constants obtained by LDA method are larger than GGA method in HfX compounds. In HfOs, the elastic constants derived from GGA method has agreed well with theoretical values from Arıkan’s [
13] and Liu’s work [
14]. Meanwhile, the elastic constants for pure Hf metal are calculated and presented with the published experimental values [
37] for comparison, where the GGA computed elastic constants are in better agreement with experimental values [
37].
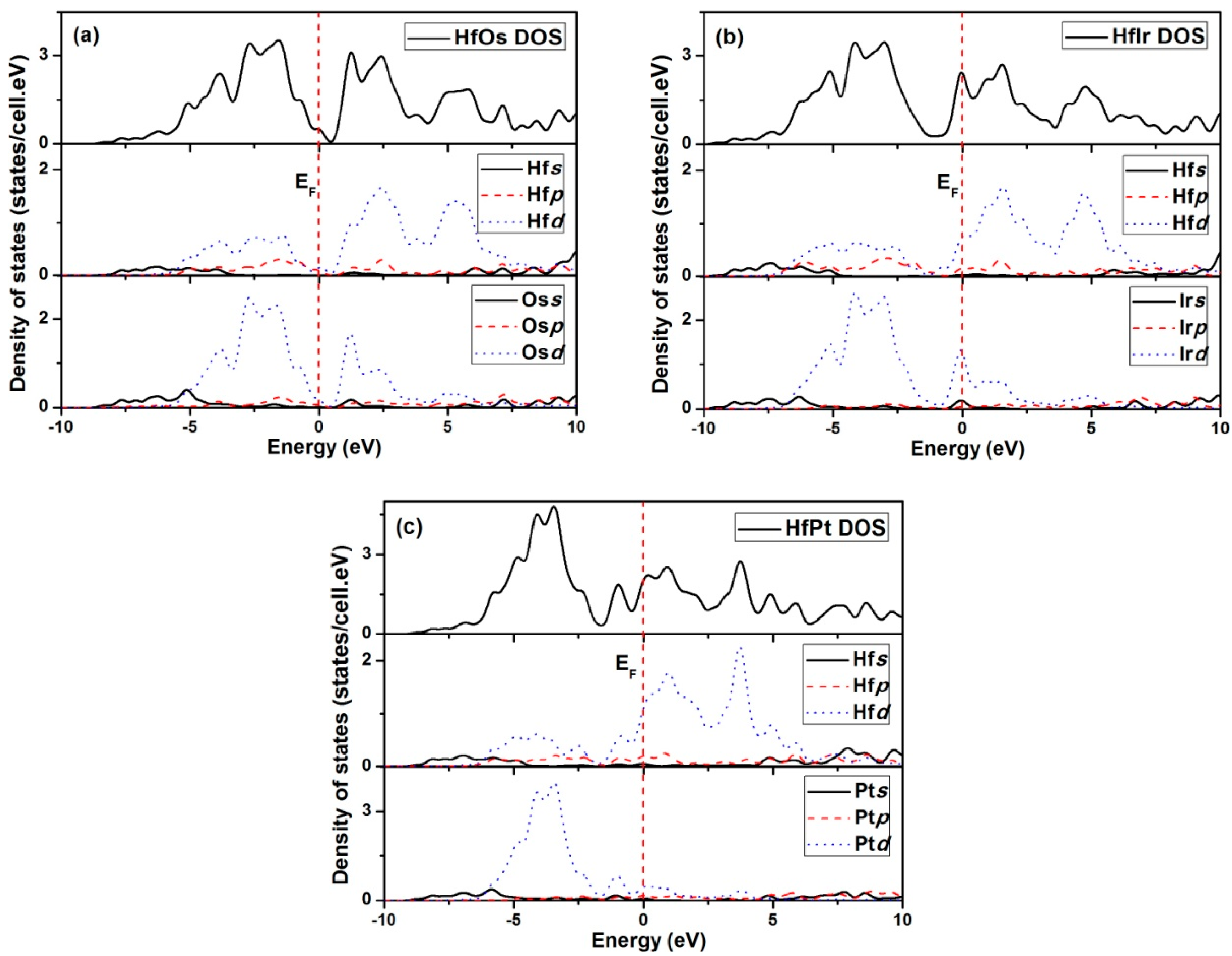
In order to analyze the elastic properties of HfX compounds, the elastic constants generated using GGA method are used as the example.
C11 is the symbol of compressive resistance along
x axis. In each compound, the calculated C
11 has the largest value, indicating the incompressible essence of the compound under the
x direction uniaxial stress [
38]. HfOs has the most incompressible ability owing to the largest
C11 (402.1 GPa). Moreover, a larger
C44 (121.0 GPa) can reflect a stronger resistance to monoclinic shear in (100) plane, suggesting HfOs also has the strongest ability to resist shear distortion in (100) plane.
Furthermore, the mechanical stability is evaluated by Born’s criteria [
39] for cubic crystals:
HfX (X = Os, Ir and Pt) compounds are all found mechanically stable at the ground state by the successful validation of Born’s criteria.
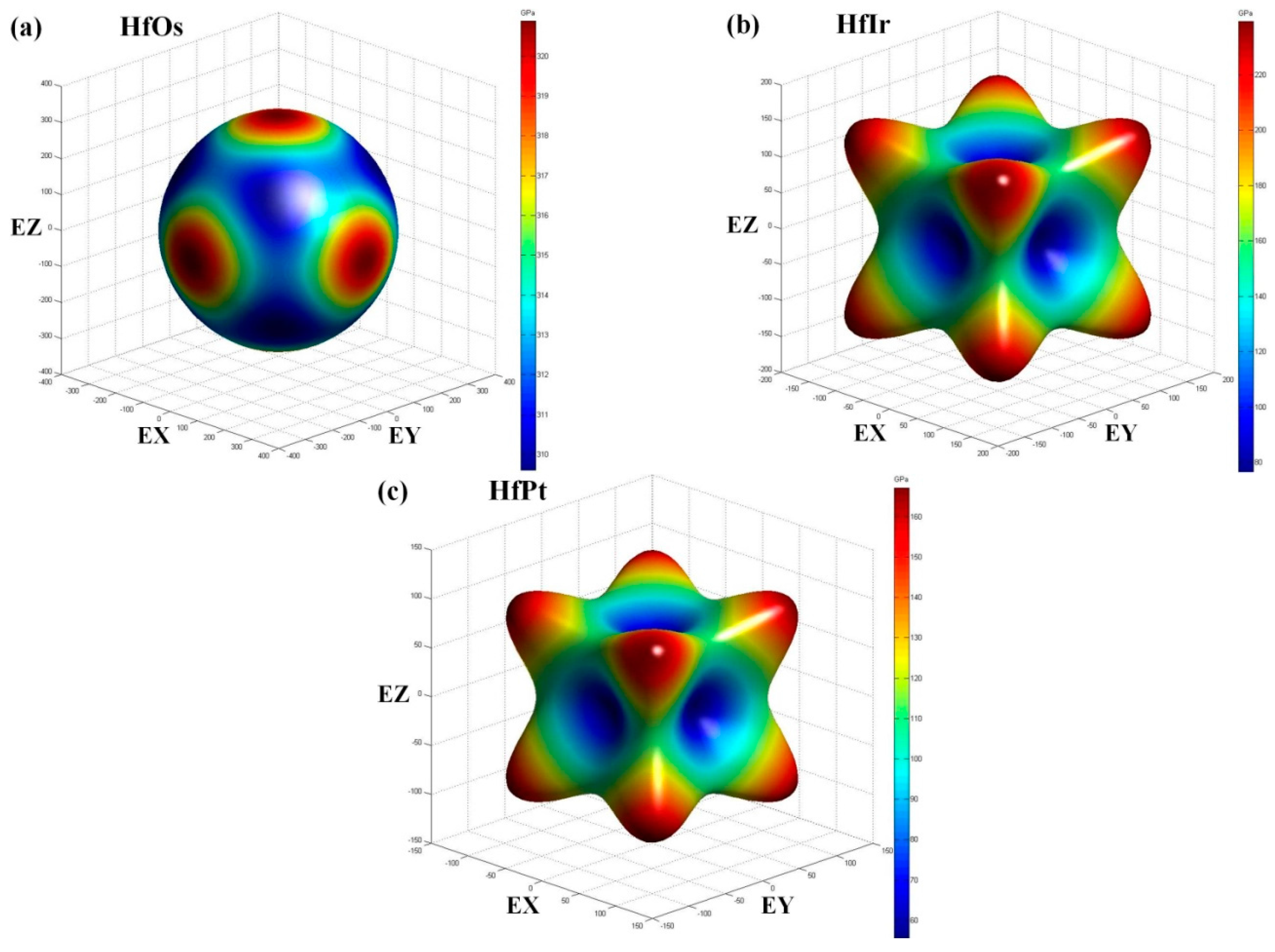
In the engineering application, the elastic properties, i.e., bulk modulus (
B), shear modulus (
G), and Young’s modulus (
E), are demanded in practice. Generally, the elastic properties can be derived from Voigt-Reuss-Hill (VRH) methods using elastic constants [
40]. For cubic crystals, the equations are expressed as following [
27,
41,
42]:
As soon as the bulk and shear moduli are achieved, Young’s modulus (
E) and Poisson’s ratio (ν) can be predicted:
The calculated elastic moduli, Poisson’s ratio and
B/
G ratio using the VRH method at the ground state are tabulated for HfX compounds in
Table 3, including the elastic moduli for pure Hf results for comparison. Overall, the elastic moduli obtained using GGA method are smaller than those provided by LDA method. Comparably, the calculated elastic moduli generated by the GGA method are in good agreement with theoretical values for HfOs. In detail, the elastic moduli are all larger than pure Hf in HfOs. However, HfIr has the similar shear and Young’s modulus over pure Hf, and HfPt owns the smaller values correspondingly. Therefore, HfOs should be a credible hardening phase in pure Hf.
Typically, the bulk modulus (
B) suggests the resistant ability against volume change under pressure of materials. In addition, the bulk moduli calculated by both methods using VRH principles are in good agreement with those provided by the EOS equations (
Table 1), confirming good self-consistency of this work. From
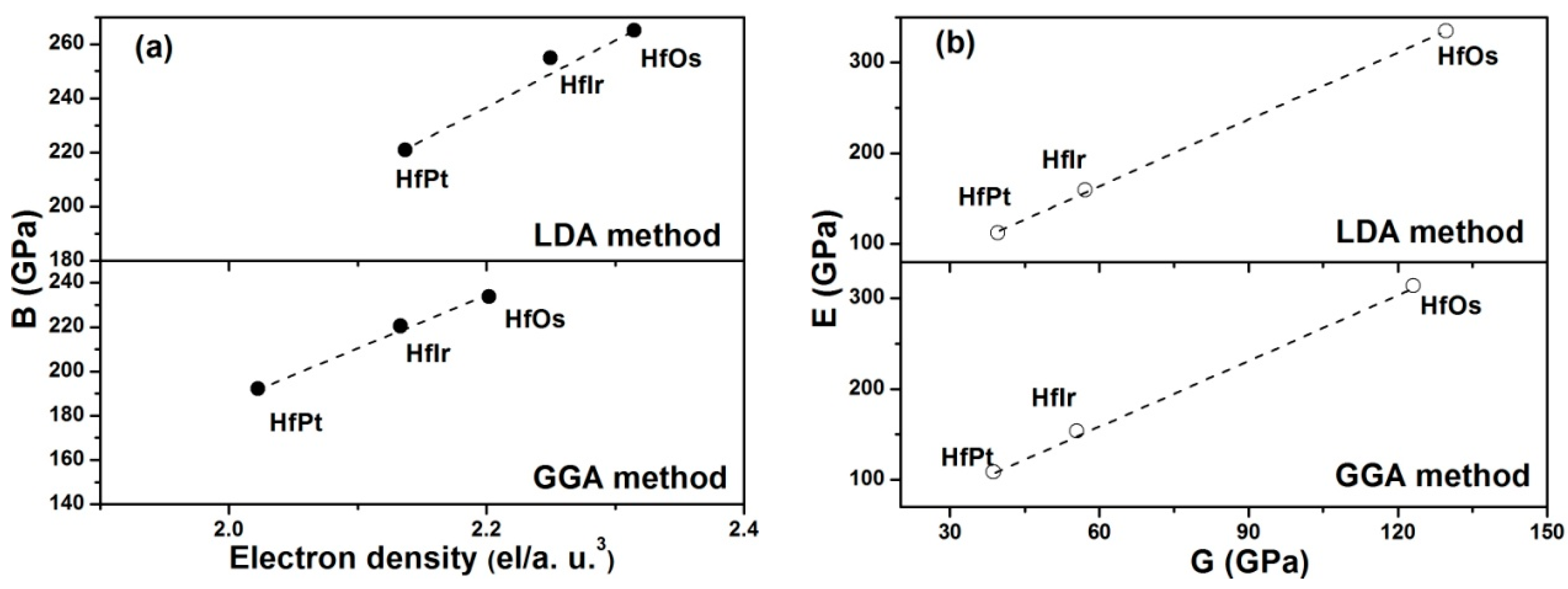
Table 3, it is seen that HfOs possesses the largest resistance to volume change by applied pressure, while HfPt has the smallest. In order to illustrate the fundamental factor on bulk moduli of HfX compounds, the relationship between electron densities and bulk moduli are constructed in
Figure 3a. Herein, the electron density (
n) is the quotient of the bonding valence (Z
B) and the volume per atom (
VM) in metal [
43]. For example, the electron density (
n) is evaluated using the following equation in HfX compounds:
where
VM(HfX) is the volume (cm
3/mol) of each HfX compound;
ZB(HfX) is the bonding valence in (el/atom) rationalized from Vegard’s law [
44]:
where the bonding valence of pure element can be found in the reference [
45].
In
Figure 3a, the correlation between the electron density and bulk modulus can be constructed using the computed values either by LDA or GGA method. In detail, the linear relationships between the electron density and bulk modulus are clearly seen in HfX compounds.
The shear modulus has reflected the resistance to reversible deformations under the shear stress [
27]. A larger shear modulus for HfOs suggests its higher resistance to reversible deformations. Young’s modulus is a symbol of the stiffness of a solid [
46]. The material with a larger Young’s modulus is stiffer. Therefore, HfOs is much stiffer than any other considered HfX compounds due to its higher Young’s modulus. Overall, Young’s modulus has linearly improved with the shear modulus in the order of HfOs > HfIr > HfPt for both LDA and GGA methods (
Figure 3b).
The
B/
G value [
47] and Poisson’s ratio (ν) [
32,
48] have determined the brittleness and ductility of the solid. A solid with
B/
G < 1.75 or ν < 0.26 is usually brittle. Otherwise, it is ductile. In
Table 3, it is found the HfPt is the most ductile compound with the largest
B/
G value and Poisson’s ratio, and HfOs is the least ductile compound owing to the smallest
B/
G value and Poisson’s ratio. Notably, the HfX compounds have similar ductile essence due to the small variations of
B/
G values and Poisson’s ratios using both GGA and LDA methods.
The hardness (
HV) is associated with the plastic and elastic properties of an intermetallic compound. The hardness can be calculated by a semi-empirical equation [
49]:
where
B and
G are the bulk modulus and shear modulus, respectively.
The hardness values (
HV) of HfX (X = Os, Ir and Pt) compounds are tabulated in
Table 3. Generally, the hardness is related with the values of
G/
B and
G that the high values of
G/
B and
G correspond to the high hardness. It is seen that the hardness value computed by GGA method is a bit larger than LDA method. Overall, the hardness values (
HV) have followed the order of HfPt < HfIr < HfOs in both GGA and LDA methods.






{kind=link}
{kind=link}
{kind=link}
{kind=link}