An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications


 , ,
, ,  , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cellular Structure Preparation
2.2. Coating of the Iron Samples
2.3. Material Characterization
2.4. Electrochemical Degradation Test
2.4.1. Potentiodynamic Polarization
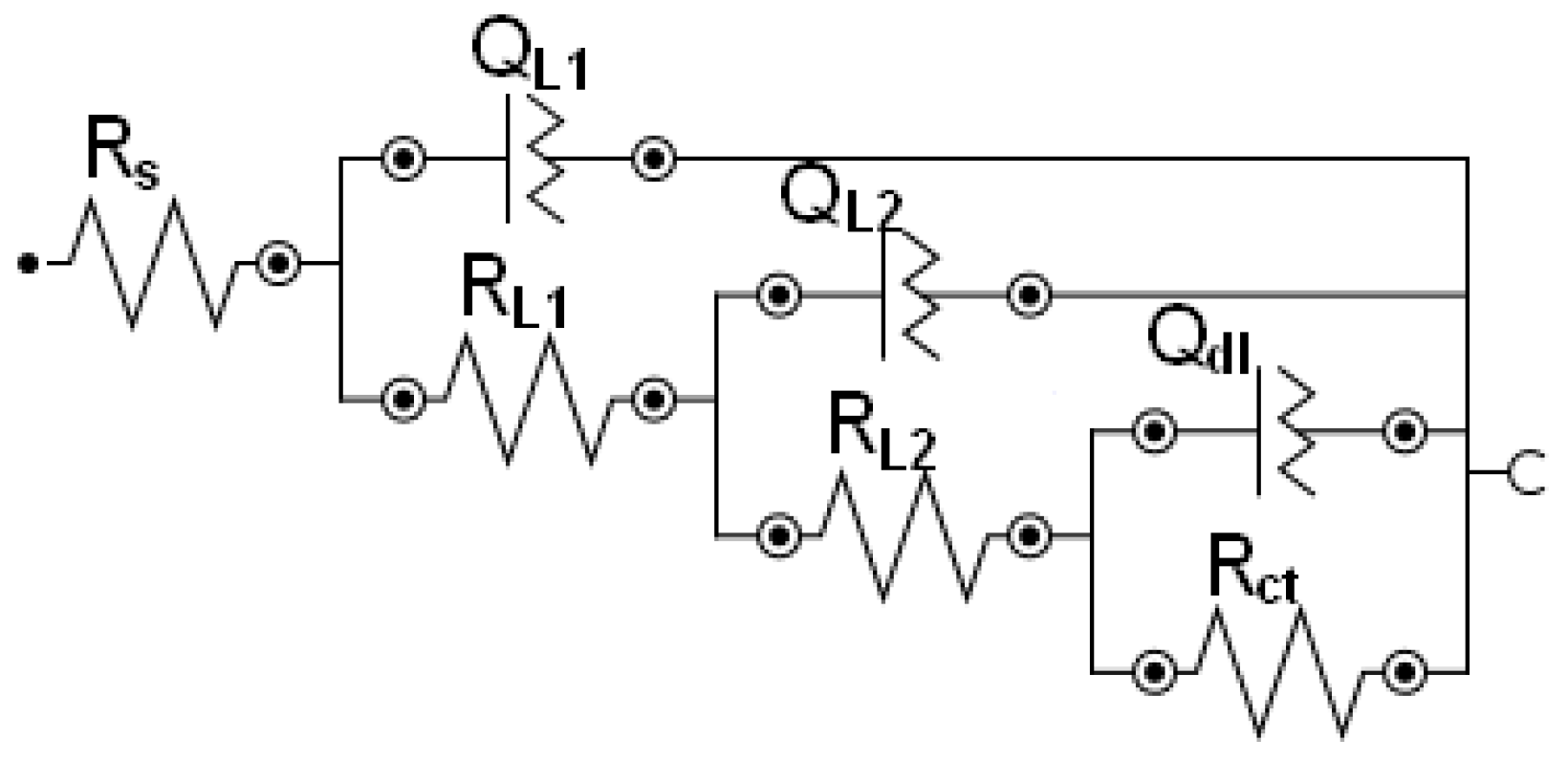
2.4.2. Electrical Impedance Spectroscopy
2.5. Static Immersion Test
3. Results
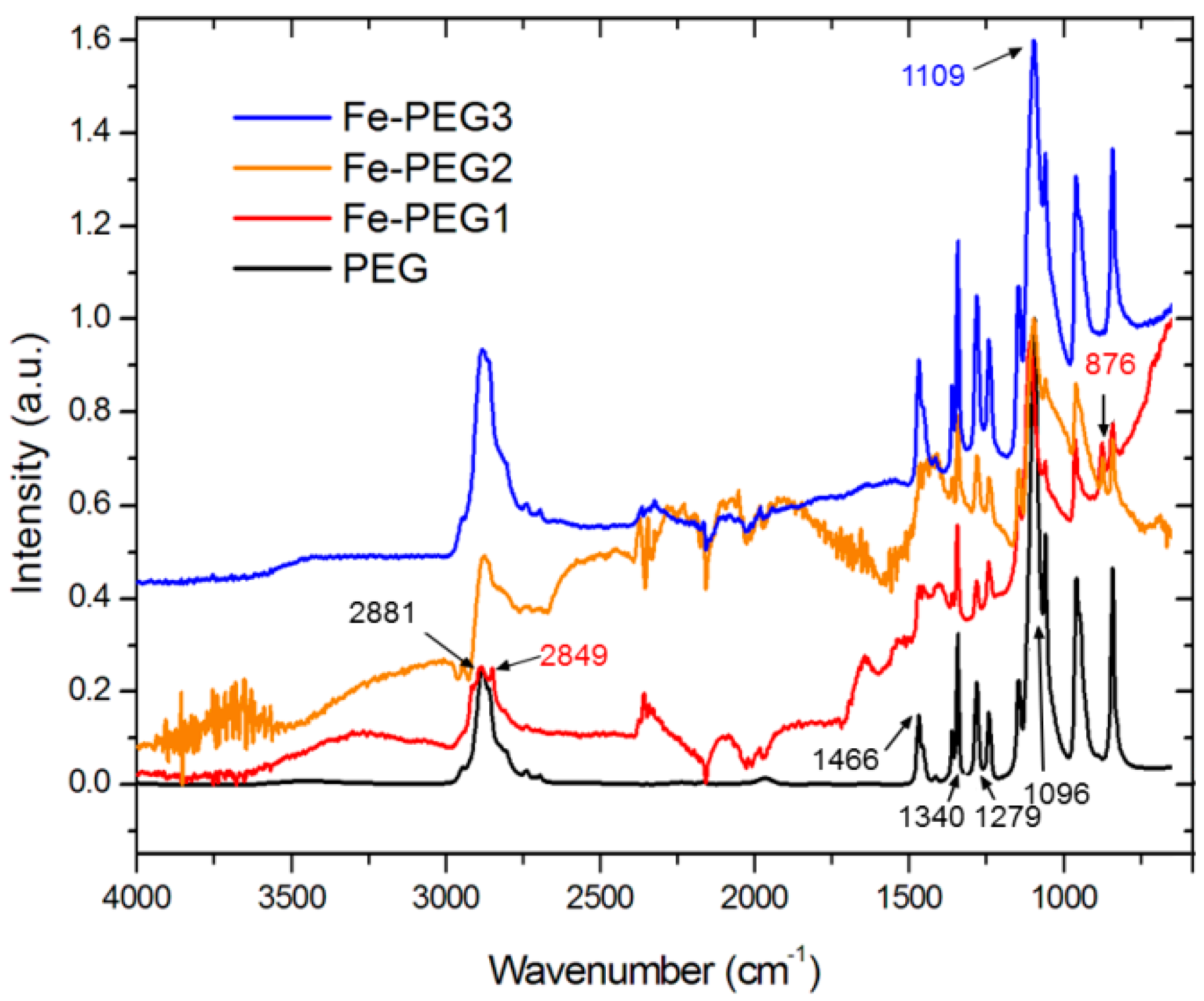
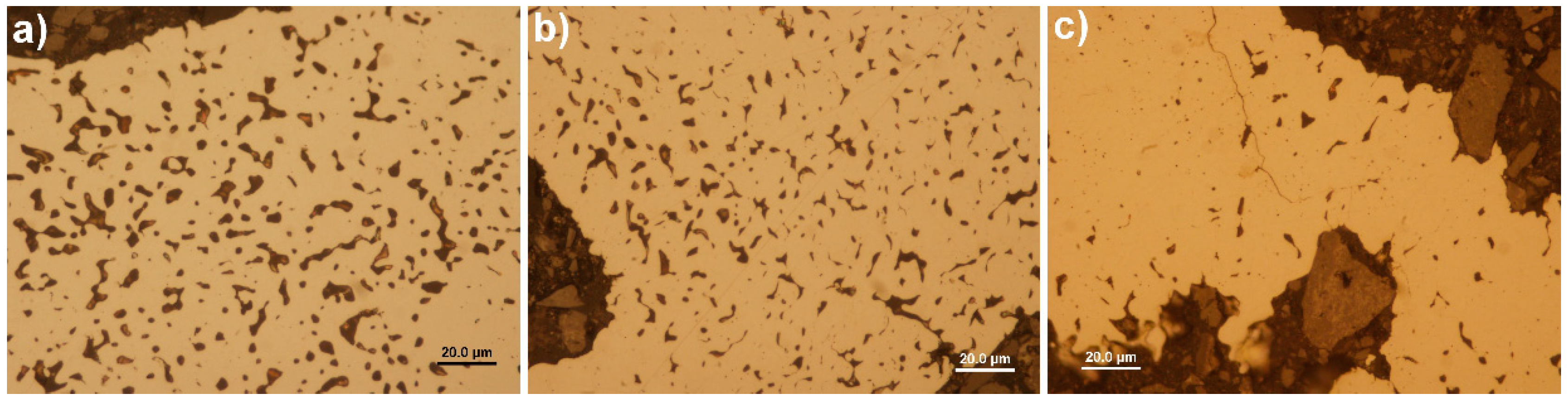
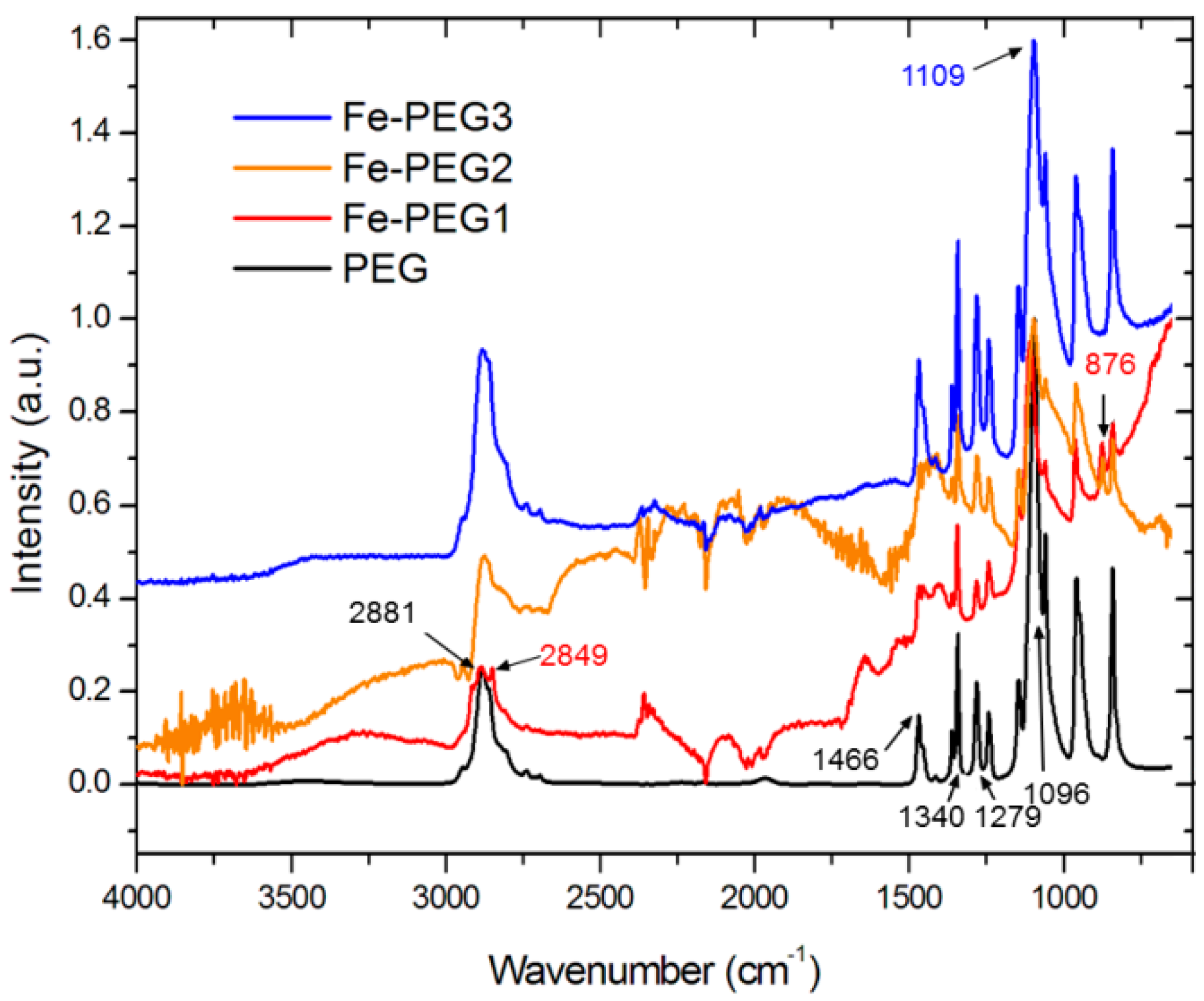
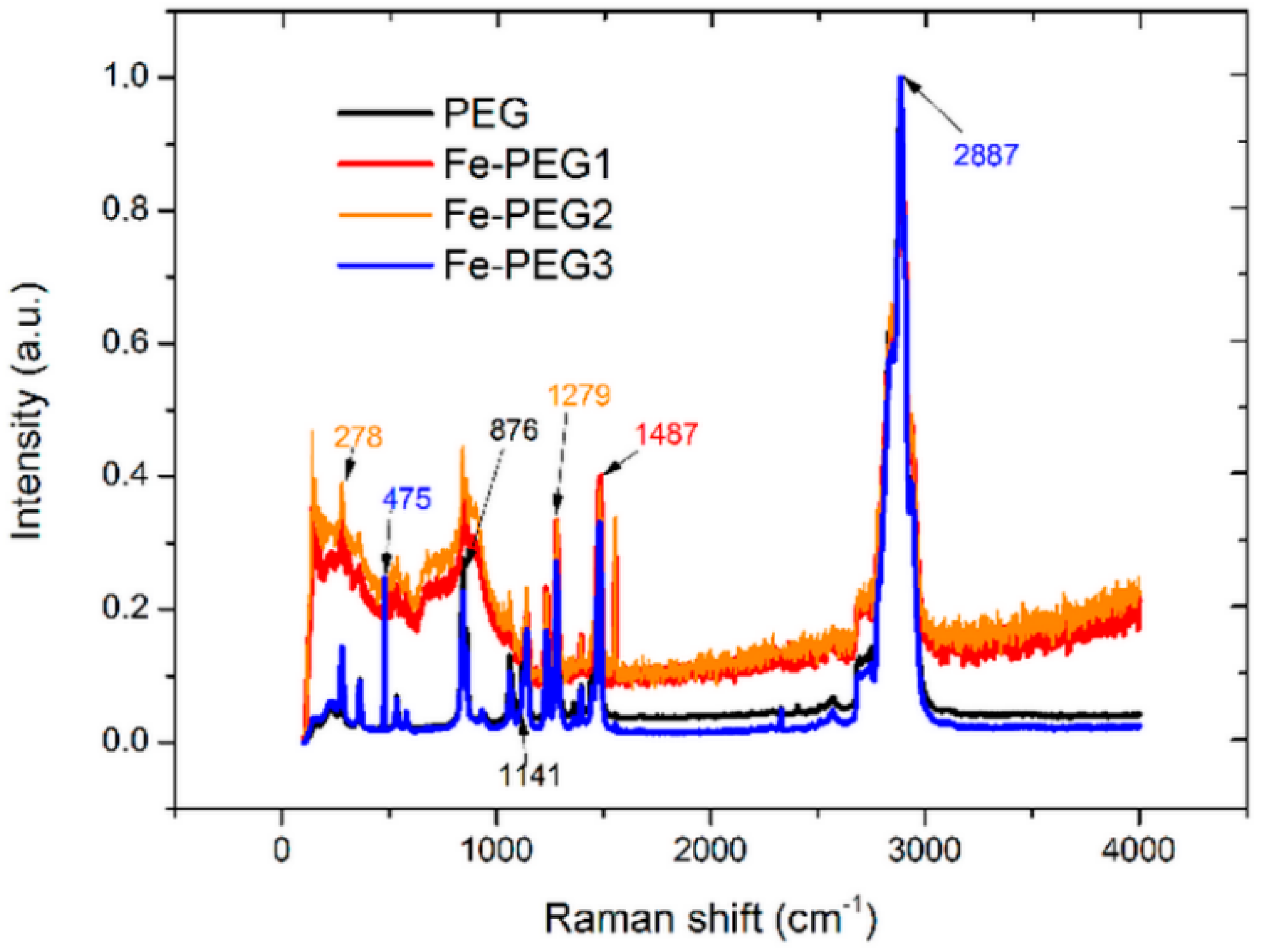
3.1. Surface Morphology and Composition
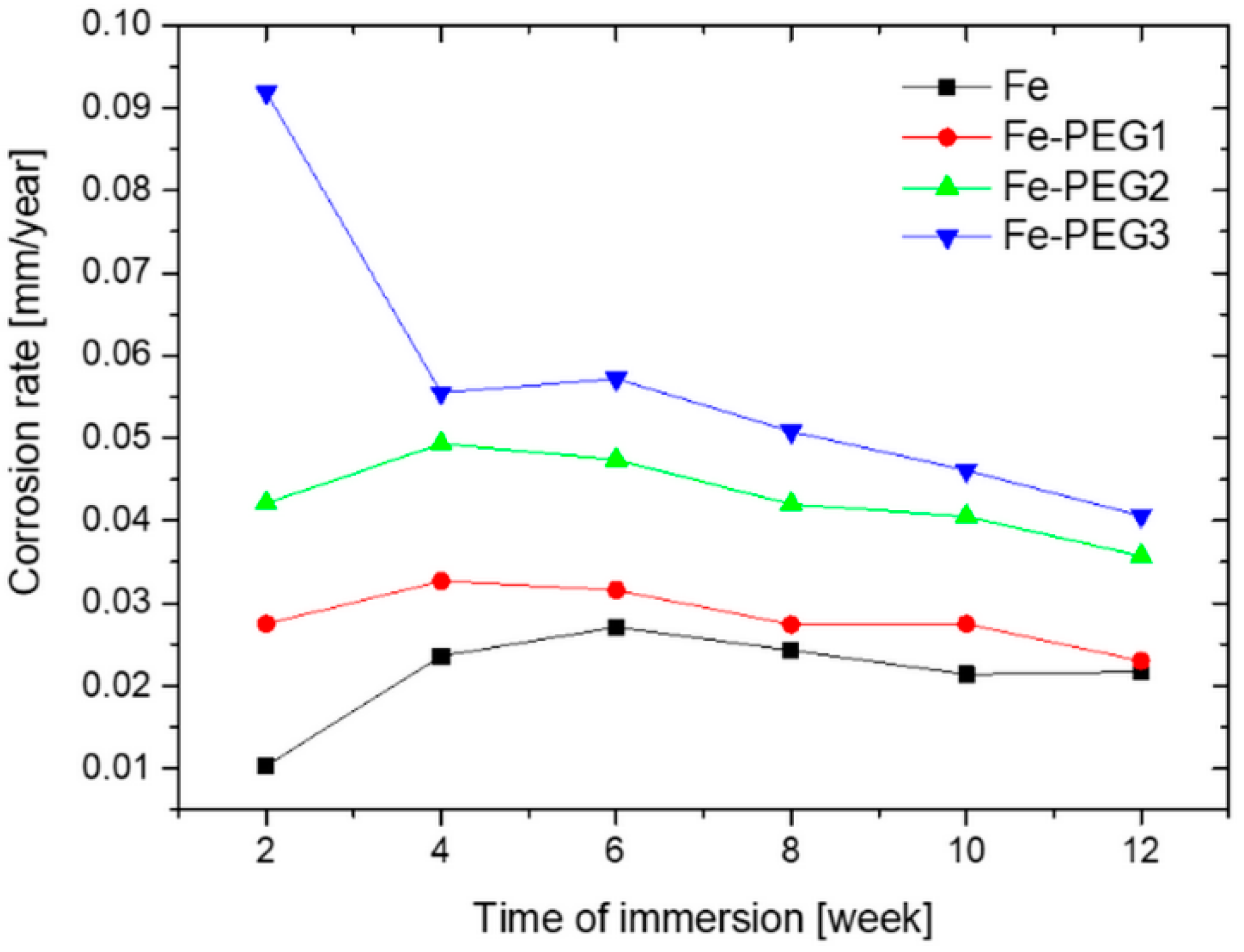
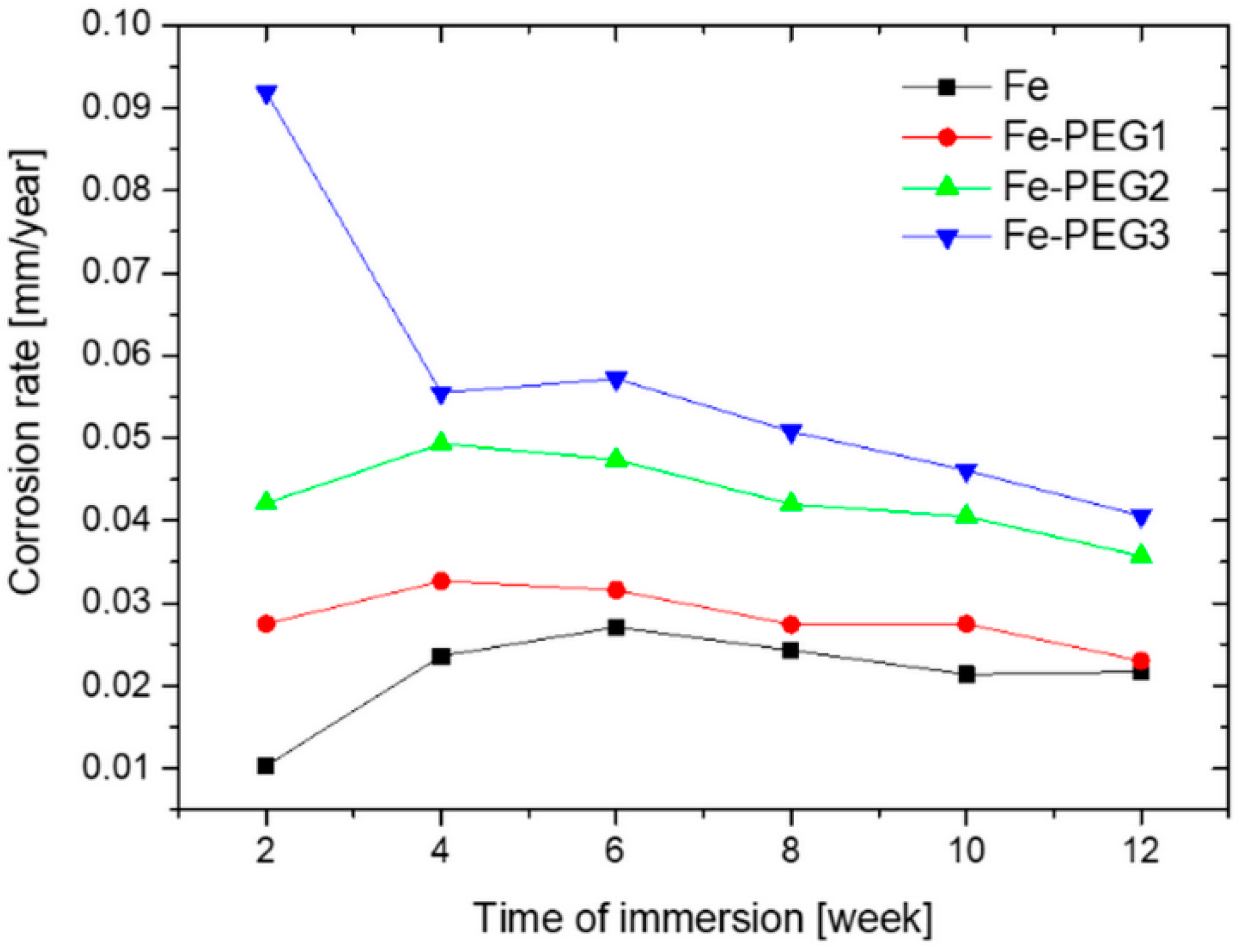
3.2. Immersion Tests
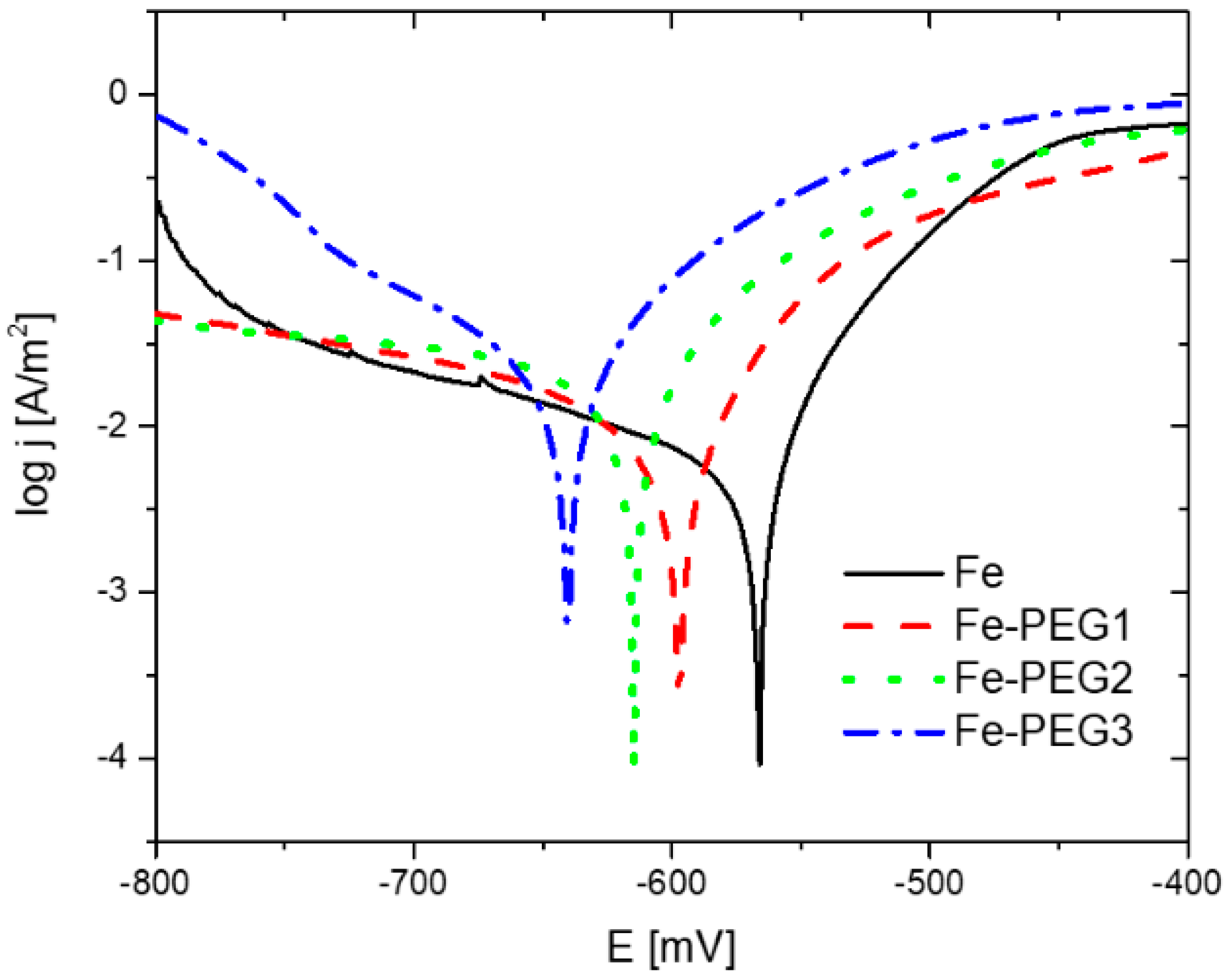
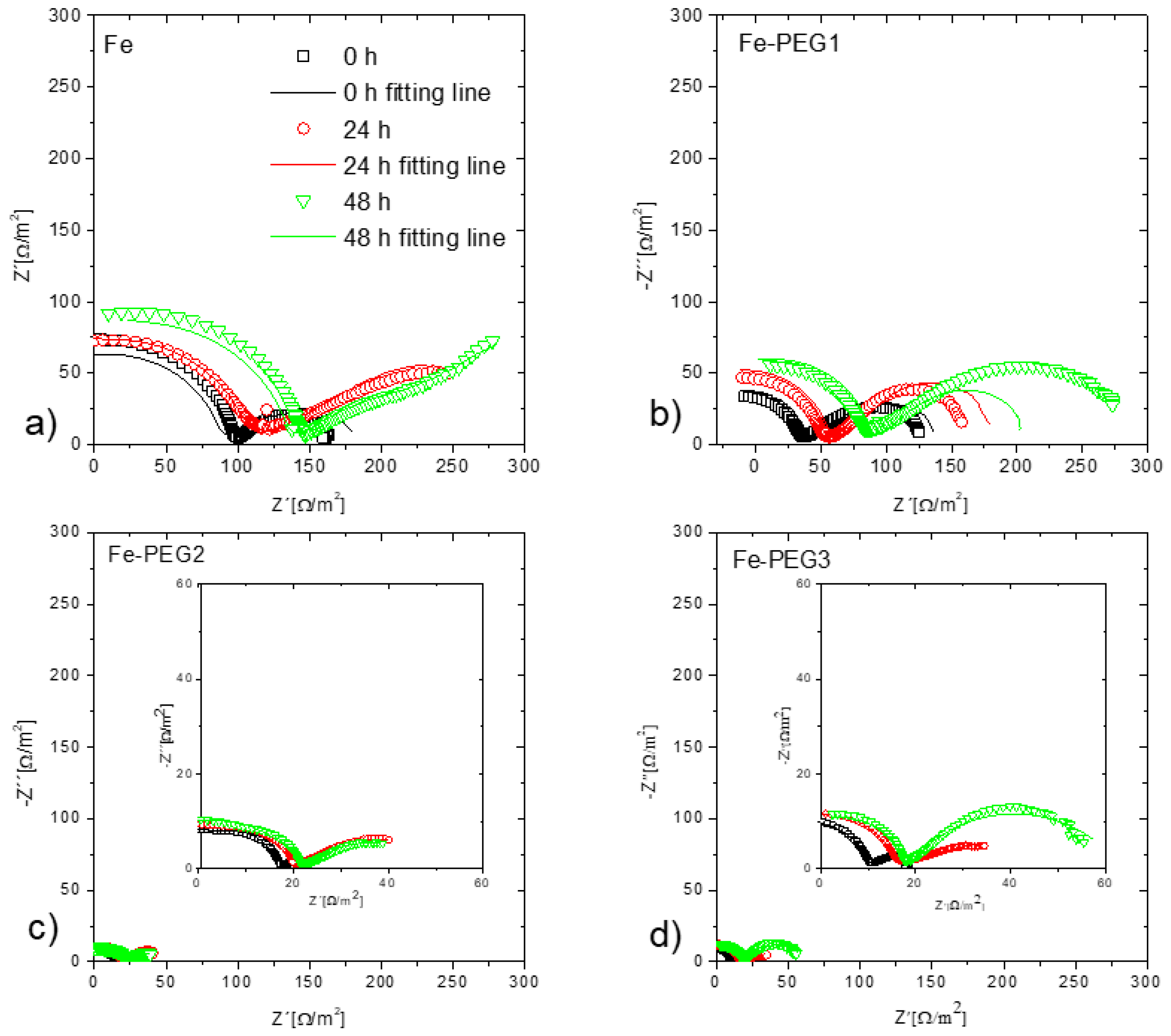
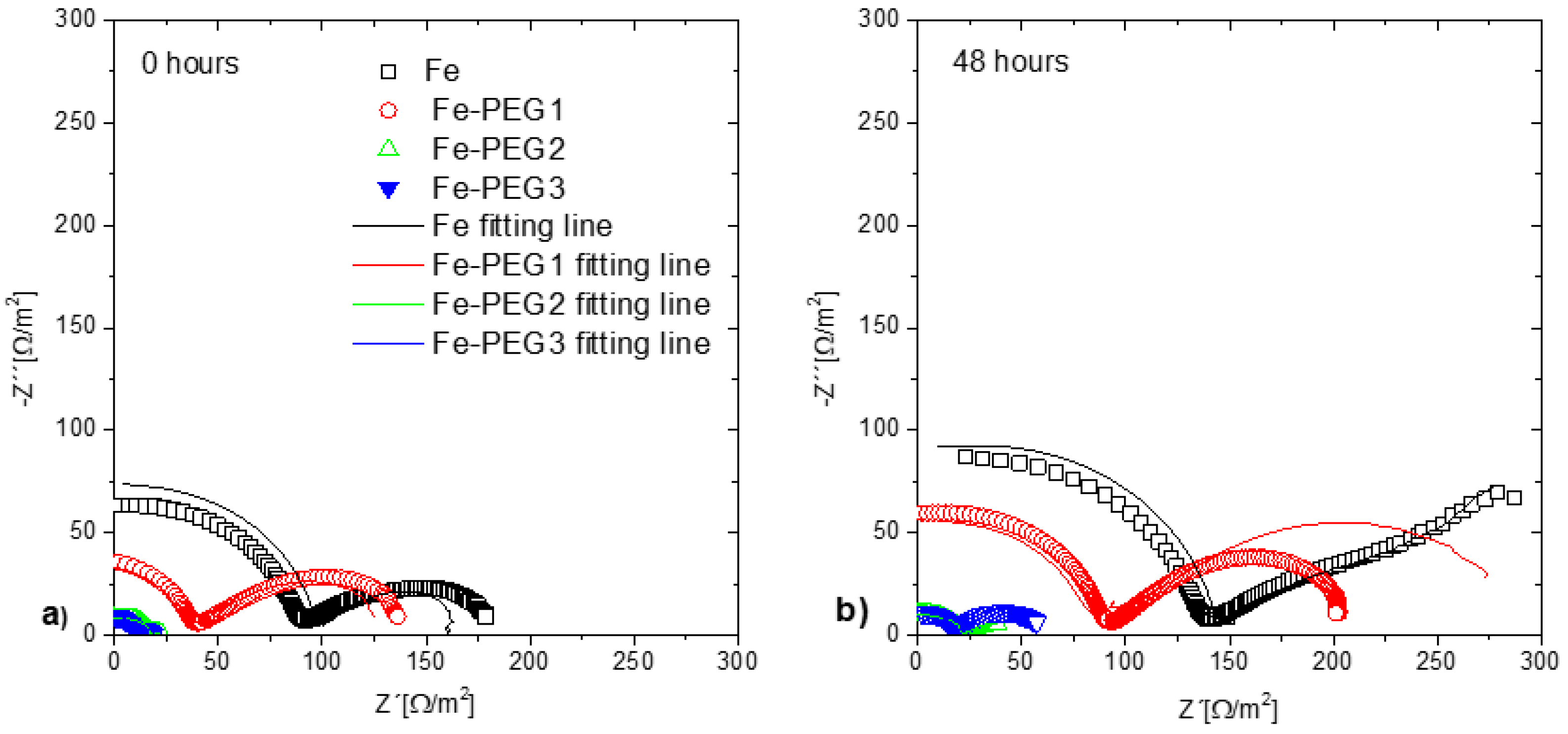
3.3. Electrochemical Corrosion Behavior
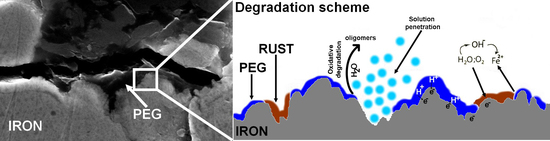
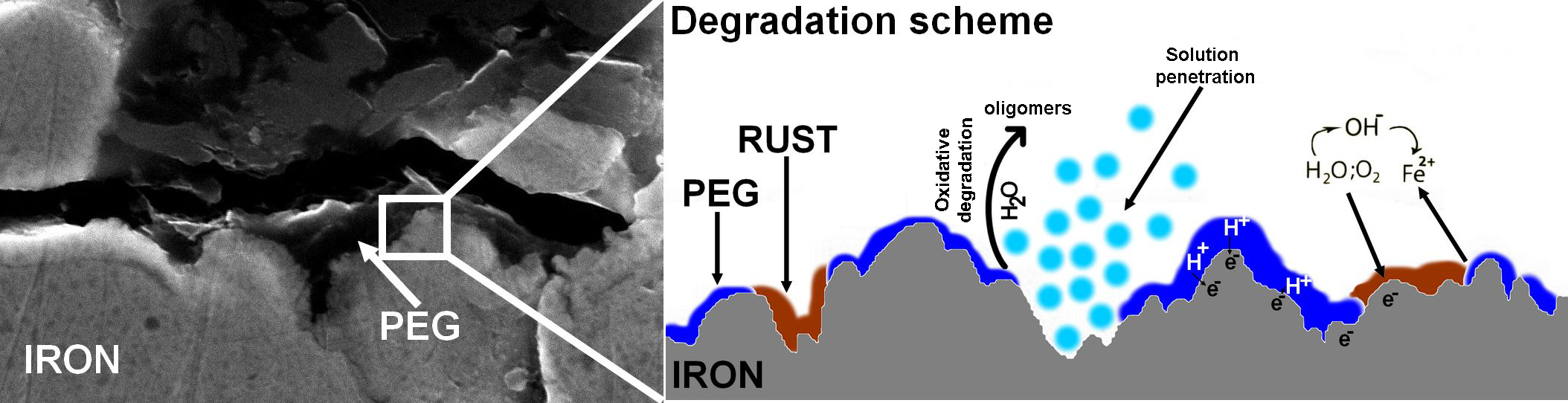
3.4. Degradation Mechanism
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
| A | exposed surface area of the sample (cm2) |
| BET | Brunauer-Emmett-Teller theory |
| CIP | carbonyl iron powder, iron powder produced from decomposition of iron(0) pentacarbonyl |
| CPE | constant phase element |
| CR | corrosion rate (in millimeters per year in thickness assuming uniform corrosion) |
| Ecorr | corrosion potential (vs. Ag/AgCl/KCl(3 mol/L)) |
| EDX | energy-dispersive X-ray (spectrometry analysis) |
| EIS | electrical impedance spectroscopy |
| EW | equivalent weight (27.92 g/eq for Fe) |
| Fe-PEG1 | sample immersed into 5 wt. % PEG solution |
| Fe-PEG2 | sample immersed into 10 wt. % PEG solution |
| Fe-PEG3 | sample immersed into 15 wt. % PEG solution |
| fmax | frequency at which the imaginary impedance component is at a maximum |
| IR | infrared (spectra) |
| jcorr | the current density (µA/cm2) |
| mi | initial mass (g) |
| mf | final mass after corrosion (g) |
| OCP | open circuit potential |
| PEG | polyethylene glycol, a sample designation |
| PEG1 | iron foam with 0.8 wt. % PEG |
| PEG2 | iron foam with 1.5 wt. % PEG |
| PEG3 | iron foam with 3.0 wt. % PEG |
| PLGA | poly(lactic-co-glycolic acid) |
| PUR | polyurethane (foam) |
| Q | value parameter of the constant phase element |
| Qdl | approximate double layer capacitance, based on a constant phase element |
| QL1 | constant phase element that corresponds to the PEG film |
| QL2 | constant phase element that corresponds to the passive film |
| Rct | charge transfer resistance |
| RL1 | pore resistance |
| RL2 | resistance of the polymer layer or corrosion products |
| Rs | solution resistance |
| ρ | density (g/cm3) |
| SBET | surface area determined by the Brunauer-Emmett-Teller method |
| SEM | scanning electron microscopy |
| t | immersion time (h) |
References
- Prasad, K.; Bazaka, O.; Chua, M.; Rochford, M.; Fedrick, L.; Spoor, J.; Symes, R.; Tieppo, M.; Collins, C.; Cao, A.; et al. Metallic Biomaterials: Current Challenges and Opportunities. Materials 2017, 10, 884. [Google Scholar] [CrossRef] [PubMed]
- Grogan, J.A.; O’Brien, B.J.; Leen, S.B.; McHugh, P.E. A corrosion model for bioabsorbable metallic stents. Acta Biomater. 2011, 7, 3523–3533. [Google Scholar] [CrossRef] [PubMed]
- Wegener, B.; Sievers, B.; Utzschneider, S.; Müller, P.; Jansson, V.; Rößler, S.; Quadbeck, P. Microstructure, cytotoxicity and corrosion of powder-metallurgical iron alloys for biodegradable bone replacement materials. Mater. Sci. Eng. B 2011, 176, 1789–1796. [Google Scholar] [CrossRef]
- Yoo, Y.W.; Park, G.J.; Lee, W.K. Surface modification of coralline scaffold for the improvement of biocompatibility and bioactivity of osteoblast. J. Ind. Eng. Chem. 2016, 33, 33–41. [Google Scholar] [CrossRef]
- Nasution, A.; Murni, N.S.; Sing, N.B.; Idris, M.H.; Hermawan, H. Partially degradable friction-welded pure iron–stainless steel 316L bone pin. J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 103, 31–38. [Google Scholar] [CrossRef] [PubMed]
- He, J.; He, F.L.; Li, D.W.; Liu, Y.L.; Liu, Y.Y.; Ye, Y.J.; Yin, D.C. Advances in Fe-based biodegradable metallic materials. RSC Adv. 2016, 6, 112819–112838. [Google Scholar] [CrossRef]
- Purnama, A.; Hermawan, H.; Couet, J.; Mantovani, D. Assessing the biocompatibility of degradable metallic materials: State-of-the-art and focus on the potential of genetic regulation. Acta Biomater. 2015, 6, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Badar, M.; Lünsdorf, H.; Evertz, F.; Rahim, M.I.; Glasmacher, B.; Hauser, H.; Mueller, P.P. The formation of an organic coat and the release of corrosion microparticles from metallic magnesium implants. Acta Biomater. 2013, 9, 7580–7589. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; He, S.; Wang, D.; Huang, D.; Zheng, T.; Wang, S.; Chen, C. In Vitro degradation and electrochemical corrosion evaluations of microarc oxidized pure Mg, Mg-Ca and Mg-Ca-Zn alloys for biomedical applications. Mater. Sci. Eng. C 2015, 47, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Rössig, C.; Angrisani, N.; Helmecke, P.; Besdo, S.; Seitz, J.M.; Welke, B.; Reifenrath, J. In Vivo evaluation of a magnesium-based degradable intramedullary nailing system in a sheep model. Acta Biomater. 2015, 25, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Böstman, O.; Pihlajamäki, H. Clinical biocompatibility of biodegradable orthopaedic implants for internal fixation: A review. Biomaterials 2000, 21, 2615–2621. [Google Scholar] [CrossRef]
- Liu, F.; Chen, C.; Niu, J.; Pei, J.; Zhang, H.; Huang, H.; Yuan, G. The processing of Mg alloy micro-tubes for biodegradable vascular stents. Mater. Sci. Eng. C 2015, 48, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Narushima, T. New-generation metallic biomaterials. In Metals for Biomedical Devices, 1st ed.; Niinomi, M., Ed.; Woodhead Publishing: Sawston, UK, 2010; pp. 355–378. ISBN 9781845699246. [Google Scholar]
- Riaz, U.; Nwaoha, C.; Ashraf, S.M. Recent advances in corrosion protective composite coatings based on conducting polymers and natural resource derived polymers. Prog. Org. Coat. 2014, 77, 743–756. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Y.; Zhou, J.; Li, H.; Chang, J.; Huan, Z. In Vitro degradation and surface bioactivity of iron-matrix composites containing silicate-based bioceramic. Bioact. Mater. 2017, 2, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Xi, T.; Zheng, Y. A review on in vitro corrosion performance test of biodegradable metallic materials. Trans. Nonferrous Met. Soc. China 2013, 23, 2283–2293. [Google Scholar] [CrossRef]
- Mouzou, E.; Paternoster, C.; Tolouei, R.; Purnama, A.; Chevallier, P.; Dubé, D.; Prima, F.; Mantovani, D. In Vitro degradation behavior of Fe-20Mn-1.2C alloy in three different pseudo-physiological solutions. Mater. Sci. Eng. C 2016, 61, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Saqaei, M.; Fathi, M.; Edris, H.; Mortazavi, V. Preparation and biocompatibility evaluation of bioactive glass–forsterite nanocomposite powder for oral bone defects treatment applications. Mater. Sci. Eng. C 2015, 56, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Matusiewicz, H. Potential release of in vivo trace metals from metallic medical implants in the human body: From ions to nanoparticles—A systematic analytical review. Acta Biomater. 2014, 10, 2379–2403. [Google Scholar] [CrossRef] [PubMed]
- Vojtěch, D.; Kubásek, J.; Čapek, J.; Pospíšilová, I. Comparative mechanical and corrosion studies on magnesium, zinc and iron alloys as biodegradable metals. Mater. Technol. 2015, 49, 877–882. [Google Scholar] [CrossRef]
- Heiden, M.; Kustas, A.; Chaput, K.; Nauman, E.; Johnson, D.; Stanciu, L. Effect of microstructure and strain on the degradation behavior of novel bioresorbable iron–manganese alloy implants. J. Biomed. Mater. Res. A 2015, 103, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Qin, L.; Qi, H.; Zhang, D.; Zhang, G.; Gao, R.; Qiu, H.; Xia, Y.; Cao, P.; Wang, X.; et al. Long-term In Vivo corrosion behavior, biocompatibility and bioresorption mechanism of a bioresorbable nitrided iron scaffold. Acta Biomater. 2017, 54, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Smith, C.; Sankar, J. Recent advances on the development of magnesium alloys for biodegradable implants. Acta Biomater. 2014, 10, 4561–4573. [Google Scholar] [CrossRef] [PubMed]
- Hampp, C.; Angrisani, N.; Reifenrath, J.; Bormann, D.; Seitz, J.M.; Meyer-Lindenberg, A. Evaluation of the biocompatibility of two magnesium alloys as degradable implant materials in comparison to titanium as non-resorbable material in the rabbit. Mater. Sci. Eng. C 2013, 33, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Witte, F.; Xi, T.; Zheng, Y.; Yang, K.; Yang, Y.; Chan, K. Recommendation for modifying current cytotoxicity testing standards for biodegradable magnesium-based materials. Acta Biomater. 2015, 21, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Halama, M.; Kreislova, K.; Lysebettens, J.V. Prediction of Atmospheric Corrosion of Carbon Steel Using Artificial Neural Network Model in Local Geographical Regions. Corrosion 2011, 67, 1–6. [Google Scholar] [CrossRef]
- Halama, M.; Tkáč, J.; Monbaliu, O.; Zhu, Y. Non-destructive technique for evaluation of degradation on solar cells. Mater. Sci. Forum 2015, 811, 3–10. [Google Scholar] [CrossRef]
- Hendry, J.A.; Pilliar, R.M. The fretting corrosion resistance of PVD surface-modified orthopedic implant alloys. J. Biomed. Mater. Res. 2001, 58, 156–166. [Google Scholar] [CrossRef]
- Razavi, M.; Fathi, M.H.; Meratian, M. Microstructure, mechanical properties and bio-corrosion evaluation of biodegradable AZ91-FA nanocomposites for biomedical applications. Mater. Sci. Eng. A 2010, 527, 6938–6944. [Google Scholar] [CrossRef]
- Alabbasi, A.; Liyanaarachichi, S.; Kannan, B.M. Polylactic acid coating on a biodegradable magnesium alloy: An in vitro degradation study by electrochemical impedance spectroscopy. Thin Solid Films 2012, 520, 6841–6844. [Google Scholar] [CrossRef]
- Mukhametkaliyev, T.; Surmeneva, M.A.; Vladescu, A.; Cotrut, C.M.; Braic, M.; Dinu, M.; Vranceanu, M.D.; Pana, I.; Mueller, M.; Surmenev, R.A. A biodegradable AZ91 magnesium alloy coated with a thin nanostructured hydroxyapatite for improving the corrosion resistance. Mater. Sci. Eng. C 2017, 75, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Champagne, S.; Trenggono, A.; Tolouei, R.; Mantovani, D.; Hermawan, H. Development and characterization of silver containing calcium phosphate coatings on pure iron foam intended for bone scaffold applications. Mater. Des. 2018, 148, 124–134. [Google Scholar] [CrossRef]
- Wen, Z.; Zhang, L.; Chen, C.; Liu, Y.; Wu, C.; Dai, C. A construction of novel iron-foam-based calcium phosphate/chitosan coating biodegradable scaffold material. Mater. Sci. Eng. C 2013, 33, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.T.; Wells, D.; Hong, D.; Lee, B.; Kuhn, H.; Kumta, P.N. Novel processing of iron–manganese alloy-based biomaterials by inkjet 3-D printing. Acta Biomater. 2013, 9, 8593–8603. [Google Scholar] [CrossRef] [PubMed]
- Čapek, J.; Kubásek, J.; Vojtěch, D.; Jablonská, E.; Lipov, J.; Ruml, T. Microstructural, mechanical, corrosion and cytotoxicity characterization of the hot forged FeMn30 (wt. %) alloy. Mater. Sci. Eng. C 2016, 58, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Čapek, J.; Vojtěch, D. Microstructural and mechanical characteristics of porous iron prepared by powder metallurgy. Mater. Sci. Eng. C 2014, 43, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zehng, Y.F.; Ruan, L. In Vitro investigation of Fe30Mn6Si shape memory alloy as potential biodegradable metallic material. Mater. Lett. 2011, 65, 540–543. [Google Scholar] [CrossRef]
- Chen, Q.; Thouas, G.A. Metallic implant biomaterials. Mater. Sci. Eng. R Rep. 2015, 87, 1–57. [Google Scholar] [CrossRef]
- Zheng, Y.F.; Gu, X.N.; Witte, F. Biodegradable metals. Mater. Sci. Eng. R Rep. 2014, 77, 1–34. [Google Scholar] [CrossRef]
- Schinhammer, M.; Gerber, I.; Hänzi, A.C.; Uggowitzer, P.J. On the cytocompatibility of biodegradable Fe-based alloys. Mater. Sci. Eng. C 2013, 33, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Törne, K.; Larsoon, M.; Norlin, A.; Weissenrieder, J. Degradation of zinc in saline solutions, plasma, and whole blood. J. Biomed. Mater. Res. Part B Appl. Biomater. 2016, 104, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Obayi, C.S.; Tolouel, R.; Paternoster, C.; Turgeon, S.; Okorie, B.A.; Obikwelu, D.O.; Cassar, G.; Buhagiar, J.; Mantovani, D. Influence of cross-rolling on the micro-texture and biodegradation of pure iron as biodegradable material for medical implants. Acta Biomater. 2015, 17, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zheng, Y.; Qin, L. Progress of biodegradable metals. Prog. Nat. Sci. 2014, 24, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; García, A.J. Biomaterial strategies for engineering implants for enhanced osseointegration and bone repair. Adv. Drug Deliv. Rev. 2015, 94, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulum, M.F.; Arafat, A.; Noviana, D.; Yusop, A.H.; Nasution, A.K.; Abdul, K.M.R.; Hermawan, H. In Vitro and In Vivo degradation evaluation of novel iron-bioceramic composites for bone implant applications. Mater. Sci. Eng. C 2014, 36, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Mahapatro, A. Bio-functional nano-coatings on metallic biomaterials. Mater. Sci. Eng. C 2015, 55, 227–251. [Google Scholar] [CrossRef] [PubMed]
- McEntire, B.J.; Bal, B.S.; Rahaman, M.N.; Chevalier, J.; Pezzotti, G. Ceramics and ceramic coatings in orthopaedics. J. Eur. Ceram. Soc. 2015, 35, 4327–4369. [Google Scholar] [CrossRef]
- Dehestani, M.; Adolfsson, E.; Stanciu, L.A. Mechanical properties and corrosion behavior of powder metallurgy iron-hydroxyapatite composites for biodegradable implant applications. Mater. Des. 2016, 109, 556–569. [Google Scholar] [CrossRef]
- Oriňaková, R.; Oriňak, A.; Kupková, M.; Hrubovčáková, M.; Bučková, L.M.; Giretová, M.; Medvecký, Ľ.; Dobročka, D.; Patruš, O.; Kaľavský, F. In Vitro degradation and cytotoxicity evaluation of iron biomaterials with hydroxyapatite film. Int. J. Electrochem. Sci. 2015, 10, 8158–8174. [Google Scholar]
- Huang, Y.; Ding, Q.; Han, S.; Yan, Y.; Pang, X. Characterisation, corrosion resistance and in vitro bioactivity of manganese-doped hydroxyapatite films electrodeposited on titanium. J. Mater. Sci. Mater. Med. 2013, 24, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Daculsi, G.; Goyenvalle, E.; Cognet, R.; Aguado, E.; Suokas, E.O. Ostoconductive properties of poly(96L/4D-lactide)/beta-tricalcium phosphate in long term animal model. Biomaterials 2011, 32, 3166–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehdipour, M.; Afshar, A. A study of the electrophoretic deposition of bioactive glass-chitosan composite coating. Ceram. Int. 2012, 38, 471–476. [Google Scholar] [CrossRef]
- Yusop, A.H.M.; Daud, N.M.; Nur, H.; Kadir, M.R.A.; Hermawan, H. Controlling the degradation kinetics of porous iron by poly (lactic-co-glycolic acid) infiltration for use as temporary medical implants. Sci. Rep. 2015, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pidhatika, B.; Rodenstein, M.; Chen, Y.; Rakhmatullina, E.; Mühlebach, A.; Acikgöz, C.; Konradi, R. Comparative stability studies of poly(2-methyl-2-oxazoline) and poly(ethylene glycol) brush coatings. Biointerphases 2012, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Alcantar, A.N.; Aydil, S.E.; Israelachvili, N.J. Polyethylene glycol-coated biocompatible surfaces. J. Biomed. Mater. Res. B Appl. Biomater. 2000, 51, 343–351. [Google Scholar] [CrossRef]
- Bauer, S.; Schmuki, P.; von der Mark, K.; Park, J. Engineering biocompatible implant surfaces: Part I: Materials and surfaces. Prog. Mater. Sci. 2013, 58, 261–326. [Google Scholar] [CrossRef]
- Oriňaková, R.; Oriňak, A.; Bučková, L.M.; Labbanczová, E.; Kupková, M.; Hrubovčáková, M.; Fedorková, A. Biodegradable Open Cell Iron Foams for Skeletal Application. Powder Metall. Prog. 2012, 12, 219–223. [Google Scholar]
- Kalantari, S.M.; Arabi, H.; Mirdamadi, S.; Mirsalehi, S.A. Biocompatibility and compressive properties of Ti-6Al-4V scaffolds having Mg element. J. Mech. Behav. Biomed. Mater. 2015, 48, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.; Kang, S.; Jeon, J.; Lee, S.H.; Kim, H.K.; Choi, H. Complex effects of alloy composition and porosity on the phase transformations and mechanical properties of powder metallurgy steels. Powder Technol. 2015, 284, 459–466. [Google Scholar] [CrossRef]
- Oriňak, A.; Oriňaková, R.; Orsagová Králová, Z.; Morovská Turoňová, A.; Kupková, M.; Hrubovčáková, M.; Džunda, R. Sintered metallic foams for biodegradable bone replacement materials. J. Porous Mater. 2014, 21, 131–140. [Google Scholar] [CrossRef]
- Hedayati, R.; Sadighi, M.; Mohammadi-Aghdam, M.; Zadpoor, A.A. Effect of mass multiple counting on the elastic properties of open-cell regular porous biomaterials. Mater. Des. 2016, 89, 9–20. [Google Scholar] [CrossRef]
- Čapek, J.; Vojtěch, D. Properties of porous magnesium prepared by powder metallurgy. Mater. Sci. Eng. C 2013, 33, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Reihaniam, M.; Asadullahpour, S.R.; Hajarpour, S.; Gheisari, K. Application of neural network and genetic algorithm to powder metallurgy of pure iron. Mater. Des. 2011, 32, 3183–3188. [Google Scholar] [CrossRef]
- Alavi, R.; Trenggono, A.; Champagne, S.; Hermawan, H. Investigation on Mechanical Behavior of Biodegradable Iron Foams under Different Compression Test Conditions. Metals 2017, 7, 202. [Google Scholar] [CrossRef]
- Zarebski, K.; Putyra, P. Iron powder-based graded products sintered by conventional method and by SPS. Adv. Powder Technol. 2015, 26, 401–408. [Google Scholar] [CrossRef]
- ASTM. Standard Test Method for Conducting Potentiodynamic Polarization Resistance Measurements; ASTM: West Conshohocken, PA, USA, 2009. [Google Scholar]
- ASTM. Standard Guide for Laboratory Immersion Corrosion Testing of Metals; ASTM: West Conshohocken, PA, USA, 2012. [Google Scholar]
- Markušová-Bučková, L.; Oriňaková, R.; Oriňak, A.; Gorejová, R.; Kupková, M.; Hrubovčáková, M.; Baláž, M.; Kovaľ, K. Static Corrosion Test of Porous Iron Material with Polymer Coating. Powder Metall. Prog. 2016, 16, 99–106. [Google Scholar] [CrossRef]
- McCafferty, E. Validation of corrosion rates measured by the Tafel extrapolation method. Corros. Sci. 2015, 47, 3202–3215. [Google Scholar] [CrossRef]
- Jüttner, K. Electrochemical impedance spectroscopy (EIS) of corrosion processes on inhomogeneous surfaces. Electrochim. Acta 1990, 35, 1501–1508. [Google Scholar] [CrossRef]
- Zhu, S.; Huang, N.; Xu, L.; Zhang, Y.; Liu, H.; Sun, H.; Leng, Y. Biocompatibility of pure iron: In Vitro assessment of degradation kinetics and cytotoxicity on endothelial cells. Mater. Sci. Eng. C 2009, 29, 1589–1592. [Google Scholar] [CrossRef]
- Moravej, M.; Purnama, A.; Fiset, M.; Couet, J.; Mantovani, D. Electroformed pure iron as a new biomaterial for degradable stents: In vitro degradation and preliminary cell viability studies. Acta Biomater. 2010, 6, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Park, Y.M.; Oh, S.K.; You, K.M.; Lee, S.H. Enhanced reduction of nitrate by supported nanoscale zero-valent iron prepared in ethanol-water solution. Environ. Technol. 2009, 30, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Bonder, J.M.; Zhang, Y.; Kiick, K.L.; Papaefthymiou, V.; Hadjipanayis, G.C. Controlling synthesis of Fe nanoparticles with polyethylene glycol. J. Magn. Magn. Mater. 2007, 311, 658–664. [Google Scholar] [CrossRef]
- Shameli, K.; Bin Ahmad, M.; Jazayeri, S.D.; Sedaghat, S.; Shabanzadeh, P.; Jahangirian, H.; Mahdavi, M.; Abdollahi, Y. Synthesis and characterization of polyethylene glycol mediated silver nanoparticles by the green method. Int. J. Mol. Sci. 2012, 13, 6639–6650. [Google Scholar] [CrossRef] [PubMed]
- Chieng, B.W.; Ibrahim, N.A.; Yunus, W.M.Z.W.; Hussein, M.Z. Poly(lactic acid)/poly(ethylene glycol) polymer nanocomposites: Effects of graphene nanoplatelets. Polymers 2013, 6, 93–104. [Google Scholar] [CrossRef]
- Seol, S.K.; Kim, D.; Jung, S.; Chang, W.S.; Kim, J.T. One-step synthesis of PEG-coated gold nanoparticles by rapid microwave heating. J. Nanomater. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- Zheng, S.; Li, X.; Zhang, Y.; Xie, Q.; Wong, Y.S.; Zheng, W.; Chen, T. PEG-nanolized ultrasmall selenium nanoparticles overcome drug resistance in hepatocellular carcinoma HepG2 cells through induction of mitochondria dysfunction. Int. J. Nanomed. 2012, 7, 3939–3949. [Google Scholar]
- Yamini, D.; Venkatasubbu, G.D.; Kumar, J.; Ramakrishnan, V. Raman scattering studies on PEG functionalized hydroxyapatite nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 117, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Samuel, A.Z.; Umapathy, S. Energy funneling and macromolecular conformational dynamics: A 2D Raman correlation study of PEG melting. Polym. J. 2014, 46, 330–336. [Google Scholar] [CrossRef]
- Zustiak, S.P.; Leach, J.B. Hydrolytically degradable poly(ethylene glycol) hydrogel scaffolds with tunable degradation and mechanical properties. Biomacromolecules 2010, 11, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Qi, H.; He, Y.; Lin, W.; Li, P.; Qin, L.; Hu, Y.; Chen, L.; Liu, Q.; Sun, H.; et al. Strategy of Metal–Polymer Composite Stent to Accelerate Biodegradation of Iron-Based Biomaterials. ACS Appl. Mater. Interfaces 2018, 10, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Tamariz, E.; Rios-Ramírez, A. Biodegradation of medical purpose polymeric materials and their impact on biocompatibility. In Biodegradation—Life of Science; Chamy, R., Ed.; InTech: London, UK, 2013; pp. 1–29. [Google Scholar]
- Vierra, A.C.; Margues, A.T.; Guedes, R.M.; Tita, V. Material model proposal for biodegradable materials. Procedia Eng. 2011, 10, 1597–1602. [Google Scholar] [CrossRef]
- Loh, X.J. The effect of pH on the hydrolytic degradation of poly (ε-caprolactone)-block-poly(ethylene glycol) copolymers. J. Appl. Polym. Sci. 2013, 127, 2046–2056. [Google Scholar] [CrossRef]
- Dumitriu, S.; Popa, V.I. Polymeric Biomaterials: Medicinal and Pharmaceutical Applications; Taylor & Francis Group: New York, NY, USA, 2013; Volume 2, ISBN 9781420094695. [Google Scholar]
- Kannan, M.B. Biodegradable polymeric coatings for surface modification of magnesium-based biomaterials. In Surface Modification of Magnesium and Its Alloys for Biomedical Applications, Volume II: Modification and Coating Techniques, 1st ed.; Narayanan, T.S., Park, I.S., Lee, M.H., Eds.; Elsevier: Cambridge, MA, USA, 2015; ISBN 978-1-78242-078-1. [Google Scholar]
- Ulbricht, J.; Jordan, R.; Luxenhofer, R. On the biodegradability of polyethylene glycol, polypeptoids and poly(2-oxazoline)s. Biomaterials 2014, 35, 4848–4861. [Google Scholar] [CrossRef] [PubMed]
- Flis, J.; Zakroczymski, T.; Kleshnya, V.; Kobiela, T.; Duś, R. Changes in hydrogen entry rate and in surface of iron during cathodic polarisation in alkaline solutions. Electrochim. Acta 1999, 44, 3989–3997. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
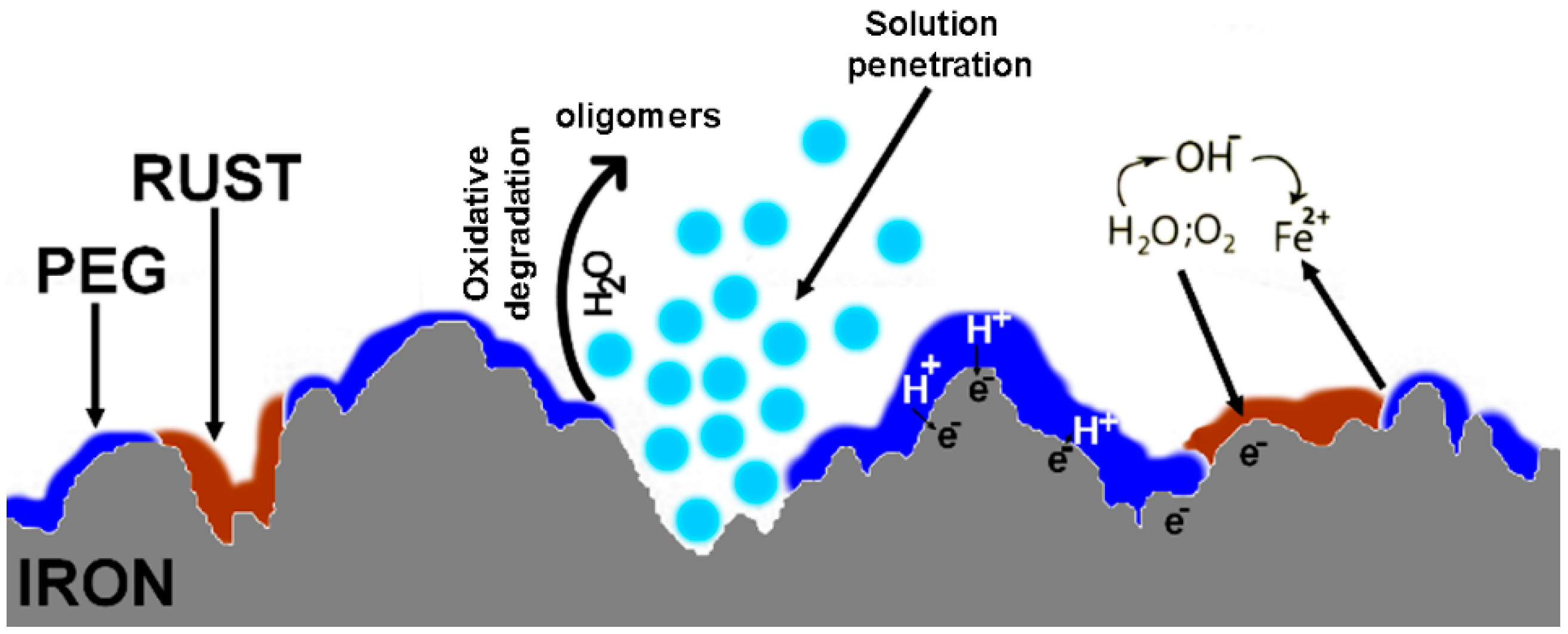{kind=link}
| Component | Concentration (g/L) |
|---|---|
| NaCl | 8.00 |
| KCl | 0.40 |
| CaCl2 | 0.14 |
| MgSO4·7H2O | 0.06 |
| NaH2PO4·2H2O | 0.06 |
| NaHCO3 | 0.35 |
| Glucose | 1.00 |
| KH2PO4 | 0.60 |
| MgCl2·6H2O | 0.10 |
| Element | Chemical Composition (wt. %) | |||
|---|---|---|---|---|
| Fe | Fe-PEG1 | Fe-PEG2 | Fe-PEG3 | |
| Fe K | 98.40 | 85.87 | 56.35 | 0 |
| C K | 0.53 | 11.42 | 35.50 | 59.32 |
| O K | 1.07 | 2.71 | 8.15 | 40.68 |
| Week | Sample Mass Loss (wt. %) | |||
|---|---|---|---|---|
| Fe | Fe-PEG1 | Fe-PEG2 | Fe-PEG3 | |
| 0 | 0 | 0 | 0 | 0 |
| 1 | 0.36 ± 0.02 | 0.82 ± 0.06 | 0.93 ± 0.06 | 1.09 ± 0.07 |
| 2 | 0.45 ± 0.04 | 2.03 ± 0.12 | 2.38 ± 0.14 | 4.04 ± 0.28 |
| 3 | 0.57 ± 0.04 | 3.22 ± 0.25 | 3.89 ± 0.30 | 4.70 ± 0.30 |
| 4 | 2.06 ± 0.16 | 4.82 ± 0.42 | 5.55 ± 0.41 | 4.88 ± 0.34 |
| 5 | 3.03 ± 0.24 | 6.26 ± 0.50 | 6.91 ± 0.56 | 5.97 ± 0.55 |
| 6 | 3.57 ± 0.33 | 6.70 ± 0.59 | 8.03 ± 0.81 | 7.54 ± 0.68 |
| 7 | 4.16 ± 0.29 | 7.25 ± 0.58 | 8.77 ± 0.70 | 8.62 ± 0.78 |
| 8 | 4.25 ± 0.41 | 8.08 ± 0.90 | 9.44 ± 1.19 | 8.93 ± 0.71 |
| 9 | 4.29 ± 0.31 | 9.30 ± 0.84 | 10.41 ± 0.92 | 9.66 ± 0.77 |
| 10 | 4.70 ± 0.60 | 10.12 ± 1.00 | 11.42 ± 1.16 | 10.13 ± 0.82 |
| 11 | 4.88 ± 0.39 | 10.13 ± 0.79 | 11.87 ± 1.07 | 10.67 ± 0.99 |
| 12 | 5.73 ± 0.63 | 10.14 ± 0.92 | 12.05 ± 1.08 | 10.72 ± 0.89 |
| Time of Immersion (weeks) | Corrosion Rate (mm/year) | |||
|---|---|---|---|---|
| Fe | Fe-PEG1 | Fe-PEG2 | Fe-PEG3 | |
| 2 | 0.0103 ± 0.0015 | 0.0275 ± 0.0038 | 0.0421 ± 0.0043 | 0.0919 ± 0.0059 |
| 4 | 0.0236 ± 0.0018 | 0.0327 ± 0.0061 | 0.0493 ± 0.0047 | 0.0555 ± 0.0064 |
| 6 | 0.0271 ± 0.0019 | 0.0316 ± 0.0050 | 0.0474 ± 0.0041 | 0.0572 ± 0.0053 |
| 8 | 0.0243 ± 0.0021 | 0.0274 ± 0.0033 | 0.0420 ± 0.0043 | 0.0508 ± 0.0046 |
| 10 | 0.0214 ± 0.0033 | 0.0275 ± 0.0040 | 0.0405 ± 0.0047 | 0.0461 ± 0.0046 |
| 12 | 0.0217 ± 0.0015 | 0.0230 ± 0.0020 | 0.0357 ± 0.0019 | 0.0406 ± 0.0034 |
| Unit | Fe | Fe-PEG1 | Fe-PEG2 | Fe-PEG3 | ||
|---|---|---|---|---|---|---|
| Tafel evaluation | Ecorr | mV | −566 ± 3 | −600 ± 5 | −614 ± 6 | −640 ± 4 |
| jcorr | μA/m2 | 0.613 ± 0.041 | 0.746 ± 0.041 | 0.859 ± 0.055 | 0.978 ± 0.077 | |
| CR | mm/year | 0.438 ± 0.029 | 0.536 ± 0.026 | 0.617 ± 0.043 | 0.703 ± 0.058 | |
| Non-Tafel evaluation | Ecorr | mV | −566 ± 2 | −598 ± 3 | −613 ± 3 | −641 ± 2 |
| jcorr | μA/m2 | 0.501 ± 0.037 | 0.6886 ± 0.034 | 0.830 ± 0.040 | 0.933 ± 0.062 | |
| CR | mm/year | 0.360 ± 0.031 | 0.494 ± 0.029 | 0.5967 ± 0.039 | 0.670 ± 0.037 | |
| Impedance Parameter | Unit | Time of Immersion | Fe | Fe-PEG1 | Fe-PEG2 | Fe-PEG3 |
|---|---|---|---|---|---|---|
| Rct | Ω m2 | 0 h | 157.08 | 75.51 | 59.95 | 22.57 |
| 24 h | 120.51 | 83.40 | 66.40 | 55.21 | ||
| 48 h | 94.84 | 85.59 | 66.45 | 57.92 | ||
| Qdl | μF/cm2 | 0 h | 10.14 | 21.09 | 26.56 | 70.55 |
| 24 h | 13.21 | 19.09 | 23.98 | 28.84 | ||
| 48 h | 16.79 | 18.60 | 23.96 | 27.49 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haverová, L.; Oriňaková, R.; Oriňak, A.; Gorejová, R.; Baláž, M.; Vanýsek, P.; Kupková, M.; Hrubovčáková, M.; Mudroň, P.; Radoňák, J.; et al. An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications. Metals 2018, 8, 499. https://doi.org/10.3390/met8070499
Haverová L, Oriňaková R, Oriňak A, Gorejová R, Baláž M, Vanýsek P, Kupková M, Hrubovčáková M, Mudroň P, Radoňák J, et al. An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications. Metals. 2018; 8(7):499. https://doi.org/10.3390/met8070499
Chicago/Turabian StyleHaverová, Lucia, Renáta Oriňaková, Andrej Oriňak, Radka Gorejová, Matej Baláž, Petr Vanýsek, Miriam Kupková, Monika Hrubovčáková, Pavol Mudroň, Jozef Radoňák, and et al. 2018. "An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications" Metals 8, no. 7: 499. https://doi.org/10.3390/met8070499
APA StyleHaverová, L., Oriňaková, R., Oriňak, A., Gorejová, R., Baláž, M., Vanýsek, P., Kupková, M., Hrubovčáková, M., Mudroň, P., Radoňák, J., Orságová Králová, Z., & Morovská Turoňová, A. (2018). An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications. Metals, 8(7), 499. https://doi.org/10.3390/met8070499