Influence of Monovalent Cations on the Efficiency of Ferrous Ion Oxidation, Total Iron Precipitation, and Adsorptive Removal of Cr(VI) and As(III) in Simulated Acid Mine Drainage with Inoculation of Acidithiobacillus ferrooxidans


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of A. ferrooxidans Cell Suspensions
2.2. Influence of Monovalent Cations on the Synthesis of Secondary Iron Minerals
2.3. Removal of Cr(VI) and As(III) by Secondary Iron Minerals
2.4. Analytical Procedures
3. Results and Discussion
3.1. Oxidation of Fe2+ during the Biogenic Formation of Secondary Iron Minerals
3.2. Total Fe Deposition Efficiency during the Biogenic Formation of Secondary Iron Minerals
3.3. Changes in the Mineral Phases during the Biogenic Formation of Secondary Iron Minerals
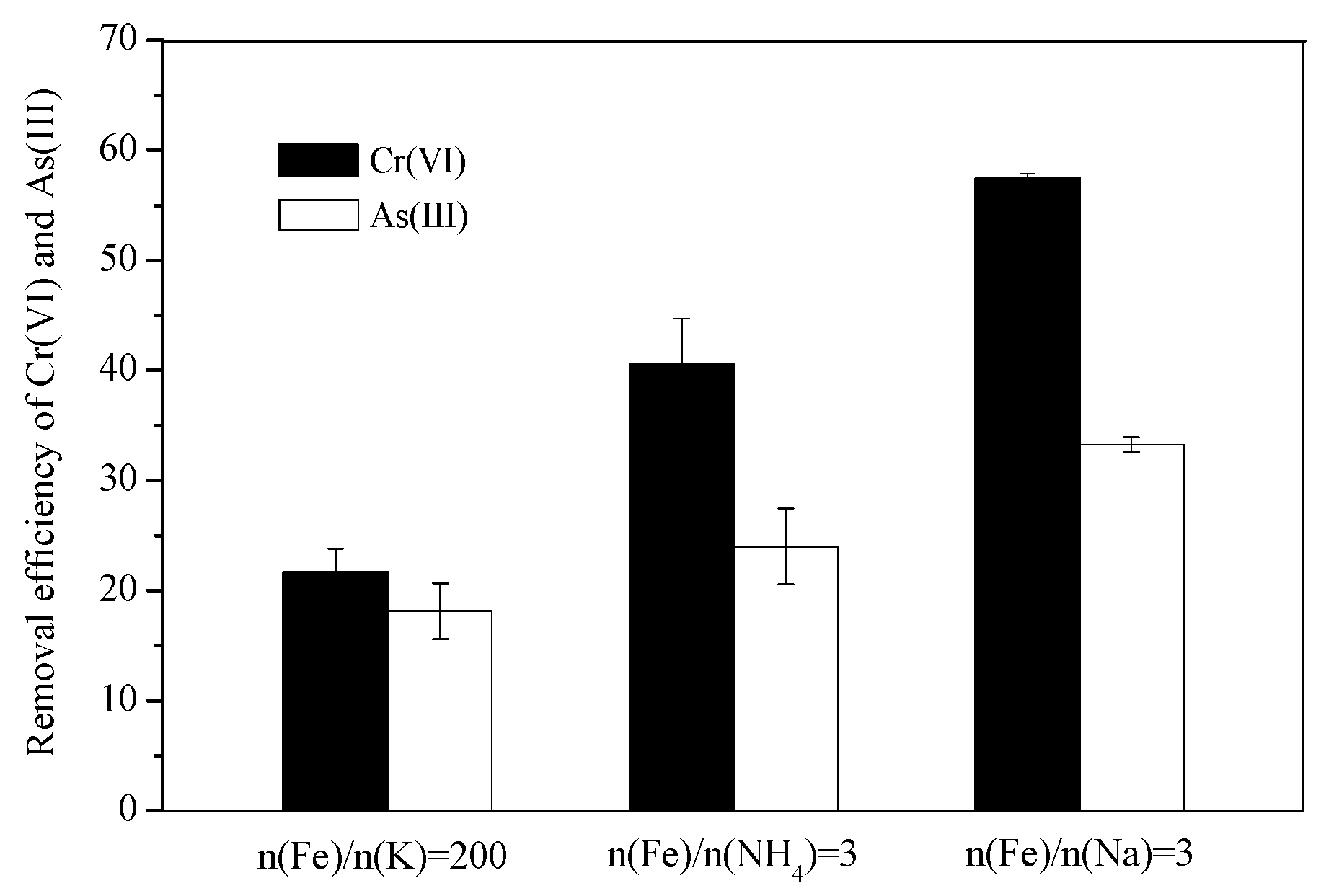
3.4. Comparison of the Cr(VI) and As(III) Removal Efficiency
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cravotta, C.A.; Trahan, M.K. Limestone drains to increase pH and remove dissolved metals from acidic mine drainage. Appl. Geochem. 1999, 14, 581–606. [Google Scholar] [CrossRef]
- Peppas, A.; Komnitsas, K.; Halikia, I. Use of organic covers for acid mine drainage control. Miner. Eng. 2000, 13, 563–574. [Google Scholar] [CrossRef]
- Sahoo, P.K.; Bhattacharyya, P.; Tripathy, S.; Equeenuddin, S.M.; Panigrahi, M.K. Influence of different forms of acidities on soil microbiological properties and enzyme activities at an acid mine drainage contaminated site. J. Hazard. Mater. 2010, 179, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Vhahangwele, M. A novel technology for neutralizing acidity and attenuating toxic chemical species from acid mine drainage using cryptocrystalline magnesite tailings. J. Water Process Eng. 2016, 10, 67–77. [Google Scholar]
- Wei, X.; Wolfe, F.A. Minerals and mine drainage. Water Environ. Res. 2013, 85, 1515–1547. [Google Scholar] [CrossRef]
- Wu, Z.L.; Zou, L.C.; Chen, J.H.; Lai, X.K.; Zhu, Y.G. Column bioleaching characteristic of copper and iron from Zijinshan sulfide ores by acid mine drainage. Int. J. Miner. Process 2016, 149, 18–24. [Google Scholar] [CrossRef]
- Akcil, A.; Koldas, S. Acid mine drainage (AMD): Causes, treatment and case studies. J. Clean. Prod. 2006, 14, 1139–1145. [Google Scholar] [CrossRef]
- España, J.S.; Pamo, E.L.; Santofimia, E.; Aduvire, O.; Reyes, J.; Barettino, D. Acid mine drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): Geochemistry, mineralogy and environmental implications. Appl. Geochem. 2005, 20, 1320–1356. [Google Scholar] [CrossRef]
- Lee, W.C.; Lee, S.W.; Yun, S.T.; Lee, P.K.; Hwang, Y.S.; Kim, S.O. A novel method of utilizing permeable reactive kiddle (PRK) for the remediation of acid mine drainage. J. Hazard. Mater. 2016, 301, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Meschke, K.; Herdegen, V.; Aubel, T.; Janneck, E.; Repke, J.U. Treatment of opencast lignite mining induced acid mine drainage (AMD) using a rotating microfiltration system. J. Environ. Chem. Eng. 2015, 4, 2848–2856. [Google Scholar] [CrossRef]
- Song, Y.W.; Wang, M.; Liang, J.R.; Zhou, L.X. High-rate precipitation of iron as jarosite by using a combination process of electrolytic reduction and biological oxidation. Hydrometallurgy 2014, 143, 23–27. [Google Scholar] [CrossRef]
- Diao, Z.; Shi, T.; Wang, S.; Huang, X.; Zhang, T.; Tang, Y.; Zhang, X.; Qiu, R. Silane-based coatings on the pyrite for remediation of acid mine drainage. Water Res. 2013, 47, 4391–4402. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.C.; Stumm, W. Acidic mine drainage: The rate-determining step. Science 1970, 167, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Valente, T.; Grande, J.A.; De, I.T.M.L.; Santisteban, M.; Cerón, J.C. Mineralogy and environmental relevance of AMD-precipitates from the Tharsis mines, Iberian Pyrite Belt (SW, Spain). Appl. Geochem. 2013, 39, 11–25. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Gan, M.; Zhang, D.; Hu, Y.H.; Chai, L.Y. The nature of schwertmannite and jarosite mediated by two strains of Acidithiobacillus ferrooxidans with different ferrous oxidation ability. Mater. Sci. Eng. C 2013, 33, 2679–2685. [Google Scholar] [CrossRef] [PubMed]
- Regenspurg, S.; Brand, A.; Peiffer, S. Formation and stability of schwertmannite in acid mining lakes. Geochim. Cosmochim. Acta 2004, 68, 1185–1197. [Google Scholar] [CrossRef]
- Loan, M.; Richmond, W.R.; Parkinson, G.M. On the crystal growth of nanoscale schwertmannite. J. Cryst. Growth 2005, 275, 1875–1881. [Google Scholar] [CrossRef]
- Barham, R.J. Schwertmannite: A unique mineral, contains a replaceable ligand, transforms to jarosites, hematites, and/or basic iron sulfate. J. Mater. Res. 1997, 12, 2751–2758. [Google Scholar] [CrossRef]
- Min, G.; Sun, S.J.; Zheng, Z.H.; Tang, H.J.; Sheng, J.R.; Zhu, J.Y.; Liu, X.X. Adsorption of Cr(VI) and Cu(II) by AlPO4 modified biosynthetic schwertmannite. Appl. Surf. Sci. 2015, 356, 986–997. [Google Scholar]
- Mihone, K.M.; Hana, F.; Sanda, R.; Lidija, C. Assessment of metal risks from different depths of jarosite tailing waste of Trepça Zinc Industry, Kosovo based on BCR procedure. J. Geochem. Explor. 2015, 148, 161–168. [Google Scholar]
- Zhang, S.L.; Jia, S.Y.; Yu, B.; Liu, Y.; Wu, S.H.; Han, X. Sulfidization of As(V)-containing schwertmannite and its impact on arsenic mobilization. Chem. Geol. 2016, 420, 270–279. [Google Scholar] [CrossRef]
- Liao, Y.H.; Liang, J.R.; Zhou, L.X. Adsorptive removal of As(III) by biogenic schwertmannite from simulated As-contaminated groundwater. Chemosphere 2011, 83, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.; Ma, S.F.; Lu, A.H. A preliminary study on the Cr(VI) removing from wastewater by precipitation of jarosite group minerals. Bull. Miner. Petrol. Geochem. 2006, 25, 335–338. [Google Scholar]
- Asta, M.P.; Cama, J.; Martínez, M.; Giménez, J. Arsenic removal by goethite and jarosite in acidic conditions and its environmental implications. J. Hazard. Mater. 2009, 171, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Dutrizac, J.E.; Kaiman, S. Synthesis and properties of jarosite-type compounds. Can. Mineral. 1976, 14, 151–158. [Google Scholar]
- Dutrizac, J.E. The effectiveness of jarosite species for precipitating sodium jarosite. JOM 1999, 51, 30–32. [Google Scholar] [CrossRef]
- Bigham, J.M.; Schwertmann, U.; Traina, S.J.; Winland, R.L.; Wolf, M. Schwertmannite and the chemical modeling of iron in acid sulfate waters. Geochim. Cosmochim. Acta 1996, 60, 2111–2121. [Google Scholar] [CrossRef]
- Gramp, J.P.; Jones, F.S.; Bigham, J.M.; Tuovinen, O.H. Monovalent cation concentrations determine the types of Fe(III) hydroxysulfate precipitates formed in bioleach solutions. Hydrometallurgy 2008, 94, 29–33. [Google Scholar] [CrossRef]
- Liao, Y.H.; Zhou, L.X.; Liang, J.R.; Xiong, H.X. Biosynthesis of schwertmannite by Acidithiobacillus ferrooxidans cell suspensions under different pH condition. Mater. Sci. Eng. C 2009, 29, 211–215. [Google Scholar] [CrossRef]
- Bai, S.Y.; Liang, J.R.; Zhou, L.X. Effects of monovalent cation and dissolved organic matter on the formation of biogenic secondary iron minerals in bioleaching system. Acta Mineral. Sinica 2011, 31, 118–125. [Google Scholar]
- Bai, S.Y.; Liang, J.R.; Zhou, L.X. Effects of iron/potassium molar ratio on mass of biogenic Fe(III) hydroxysulfate precipitates in the FeSO4-K2SO4-H2O system and their environmental implications. Acta Sci. Circumst. 2015, 35, 476–483. [Google Scholar]
- Silverman, M.P.; Lundgren, D.G. Studies on the chemoautotrophic iron bacterium Ferrobacillus ferrooxidans. І. An improved medium and a harvesting procedure for securing high cell yields. J. Bacteriol. 1959, 77, 642–647. [Google Scholar] [PubMed]
- Wang, S.M.; Zhou, L.X. A renovated approach for increasing colony count efficiency of Thiobacillus ferrooxidans and Thiobacillus thiooxidans: Double-layer plates. Acta Sci. Circumst. 2005, 25, 1418–1420. [Google Scholar]
- Csonka, L.N. Physiological and genetic responses of bacteria to osmotic stress. Microbiol. Rev. 1989, 53, 121–147. [Google Scholar] [PubMed]
- Wang, Q.L.; Qiu, G.Z.; Yu, R.L.; Hu, E.M.; Zhang, H.C.; Xiong, X. Effects of coexisting ions on activity of Thiobacillus ferrooxidans in in-situ leaching of uranium. China Min. Mag. 2015, 24, 116–119. [Google Scholar]
- Wu, Q.T. Study on treatment technique of preserved szechuan pickle wastewater with high salinity. Master’s Thesis, Chongqing University, Chongqing, China, 2007. [Google Scholar]
- Song, Y.W.; Wang, H.R.; Cao, Y.X.; Zhu, Y.H.; Zhou, L.X. Effect of NH4+ on the formation of secondary iron minerals and the removal of heavy metals. China Environ. Sci. 2018, 38, 2116–2123. [Google Scholar]
- Suzuki, I.; Lee, D.; Mackay, B.; Harahuc, L.; Oh, J.K. Effect of various ions, pH, and osmotic pressure on oxidation of elemental sulfur by Thiobacillus thiooxidans. Appl. Environ. Microbiol. 1999, 65, 5163–5171. [Google Scholar] [PubMed]
- Sasaki, K.; Konno, H. Morphology of jarosite-group compounds precipitated from biologically and chemically oxidized Fe ions. Can. Mineral. 2000, 38, 45–56. [Google Scholar] [CrossRef]
- Regenspurg, S.; Peiffer, S. Arsenate and chromate incorporation in schwertmannite. Appl. Geochem. 2005, 20, 1226–1239. [Google Scholar] [CrossRef]
- Bigham, J.M.; Schwertmann, U.; Carlson, L. A poorly crystallized xyhydoxysulfate of iron formed by bacterial oxidation of Fe(II) in acid mine waters. Geochim. Cosmochim. Acta 1990, 54, 2743–2758. [Google Scholar] [CrossRef]
- Duquesne, K.; Lebrun, S. Immobilization of arsensite and ferric iron by Acidithiobacillus ferrooxidans and its relevance to acid mine drainage. Appl. Environ. Microbiol. 2003, 69, 6165–6173. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, J.; Persson, P.; Sjöberg, S.; Lövgren, L. Schwertmannite precipitated from acid mine drainage: Phase transformation, sulfate release and surface properties. Appl. Geochem. 2005, 20, 179–191. [Google Scholar] [CrossRef]
- Šubrt, J.; Boháček, J.; Štengl, V.; Grygar, T.; Bezdička, P. Uniform particles with a large surface area formed by hydrolysis of Fe2(SO4)3 with urea. Mater. Res. Bull. 1999, 34, 905–914. [Google Scholar] [CrossRef]
- Sun, H.F.; Zhao, F.H.; Cong, Z.Y.; Yue, M.; Ren, D.Y. The mineral schwertmannite found in China and its characteristics. Acta Mineral. Sinica 2006, 26, 38–42. [Google Scholar]
- Dold, B. Dissolution kinetics of schwertmannite and ferrihydrite in oxidized mine samples and their detection by differential X-ray diffraction (DXRD). Appl. Geochem. 2003, 18, 1531–1540. [Google Scholar] [CrossRef]
- Liu, F.W.; Bu, Y.S.; Tian, G.J.; Cui, C.H.; Zhou, L.X. Influence of temperature and pH on dissolution behavior of biogenic schwertmannite in acidic environment and the adsorption of Cu2+. Acta Sci. Circumst. 2013, 33, 2445–2451. [Google Scholar]
- Liu, F.W.; Zhou, J.; Zhang, S.S.; Liu, L.L.; Zhou, L.X.; Fan, W.H. Schwertmannite synthesis through ferrous ion chemical oxidation under different H2O2 supply rates and its removal efficiency for arsenic from contaminated groundwater. PLoS ONE 2015, 10, e0138891. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Wang, H.; Yang, J.; Cao, Y. Influence of Monovalent Cations on the Efficiency of Ferrous Ion Oxidation, Total Iron Precipitation, and Adsorptive Removal of Cr(VI) and As(III) in Simulated Acid Mine Drainage with Inoculation of Acidithiobacillus ferrooxidans. Metals 2018, 8, 596. https://doi.org/10.3390/met8080596
Song Y, Wang H, Yang J, Cao Y. Influence of Monovalent Cations on the Efficiency of Ferrous Ion Oxidation, Total Iron Precipitation, and Adsorptive Removal of Cr(VI) and As(III) in Simulated Acid Mine Drainage with Inoculation of Acidithiobacillus ferrooxidans. Metals. 2018; 8(8):596. https://doi.org/10.3390/met8080596
Chicago/Turabian StyleSong, Yongwei, Heru Wang, Jun Yang, and Yanxiao Cao. 2018. "Influence of Monovalent Cations on the Efficiency of Ferrous Ion Oxidation, Total Iron Precipitation, and Adsorptive Removal of Cr(VI) and As(III) in Simulated Acid Mine Drainage with Inoculation of Acidithiobacillus ferrooxidans" Metals 8, no. 8: 596. https://doi.org/10.3390/met8080596