Comprehensive Analysis of Prokaryotes in Environmental Water Using DNA Microarray Analysis and Whole Genome Amplification

Abstract
:1. Introduction
2. Results and Discussion
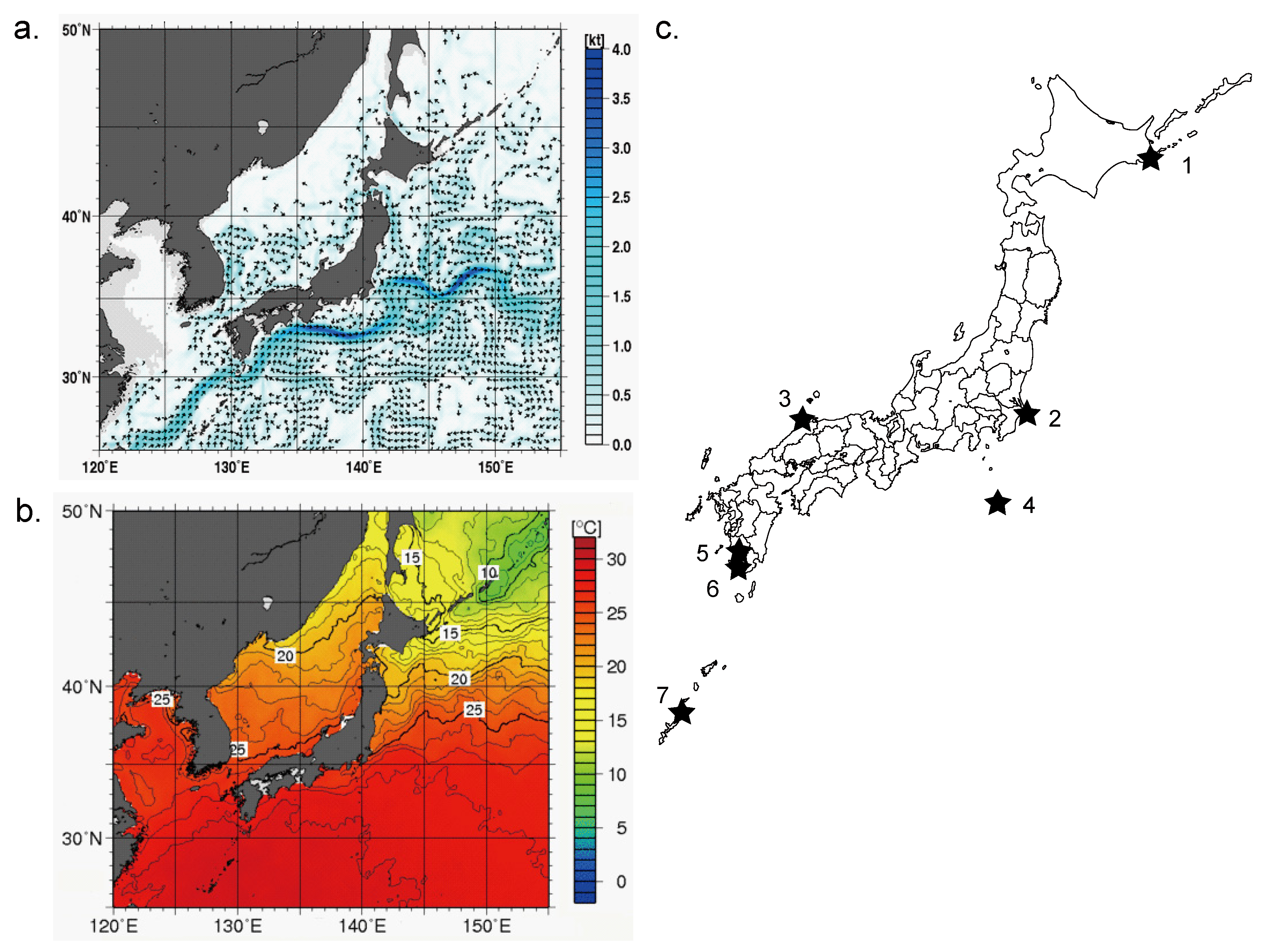
2.1. Sampling of Environmental Water
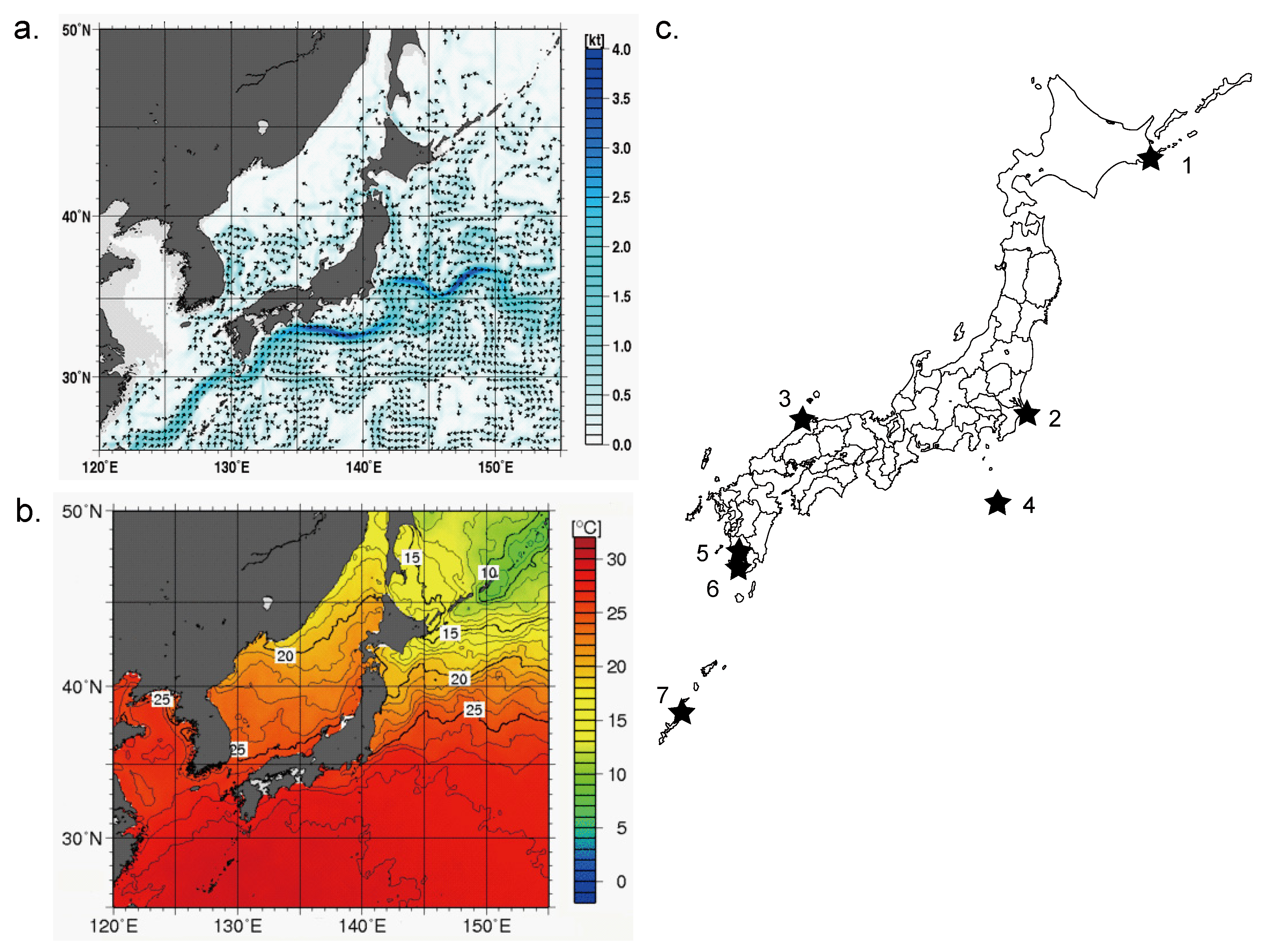

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sampling site | Salinity | Sampling | Temperature (°C) | pH | DNA recovery (μg) |
|---|---|---|---|---|---|---|
| 1 | Nemuro | Marine | July 28 | 13.5 | 7.9 | 1.10 |
| 2 | Tonegawa river | Estuarine water | May 23 | 21.0 | 8.0 | 5.60 |
| 3 | Shimanekaka | Marine | August 18 | 27.0 | 8.2 | 6.20 |
| 4 | Hachijo | Marine | August 3 | 28.0 | 8.3 | 0.12 |
| 5 | Kagoshima | Marine | September 6 | 29.9 | 8.2 | 0.24 |
| 6 | Ikeda lake | Fresh water | August 3 | 28.8 | 8.3 | 0.96 |
| 7 | Yoron | Marine | September 10 | 29.5 | 8.0 | 0.66 |
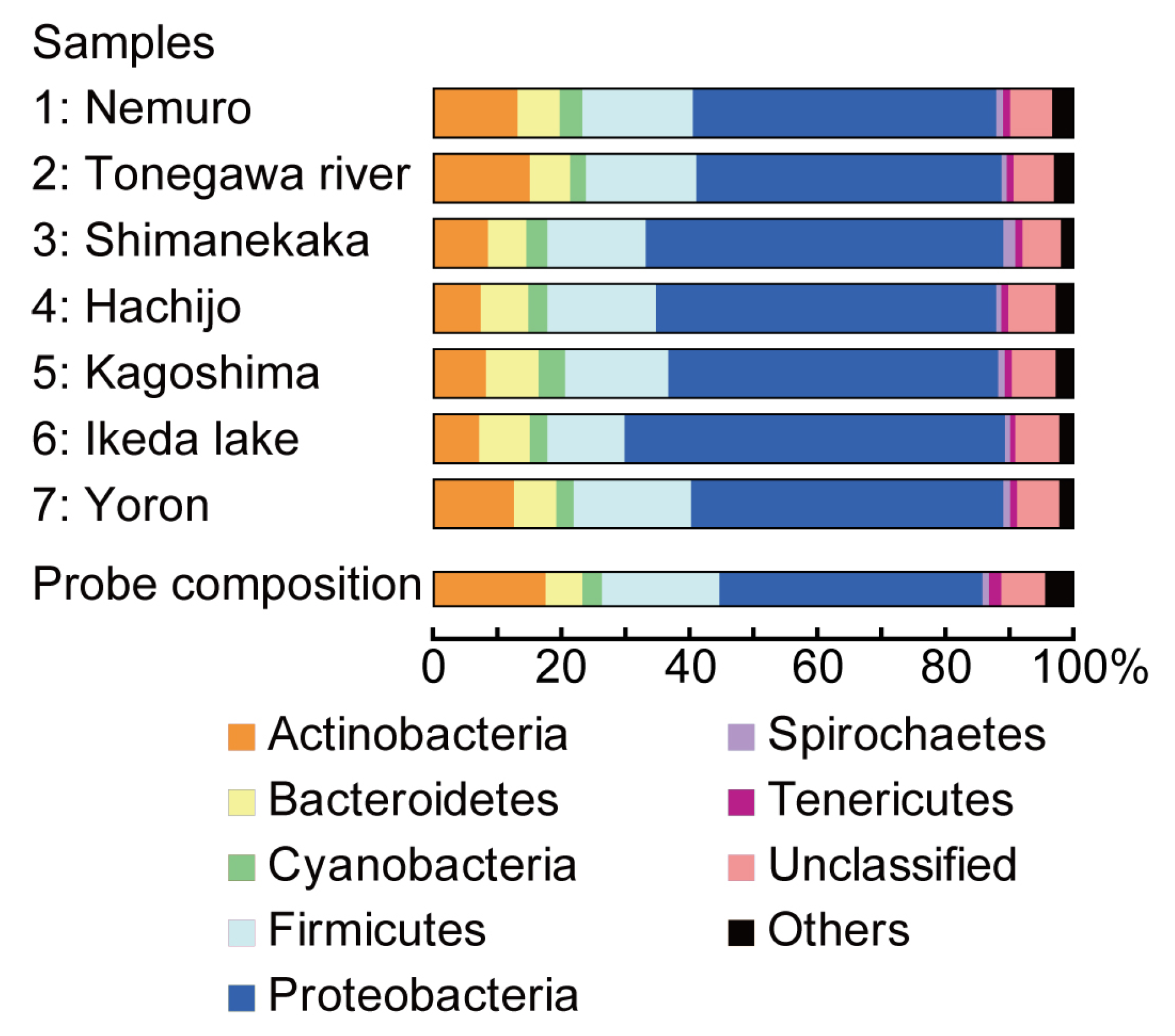
2.2. Taxonomic Composition in Each Water Sample

2.3. Detection of Pathogenic Bacteria
| Species | Sample number | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Aeromonas sobria | − | + | + | + | + | + | + |
| Citrobacter freundii | − | − | + | + | + | + | + |
| Clostridium perfringens | + | + | + | + | + | + | + |
| Mycobacterium gordonae | − | − | − | − | − | − | + |
| Mycobacterium marinum | − | + | − | − | − | − | − |
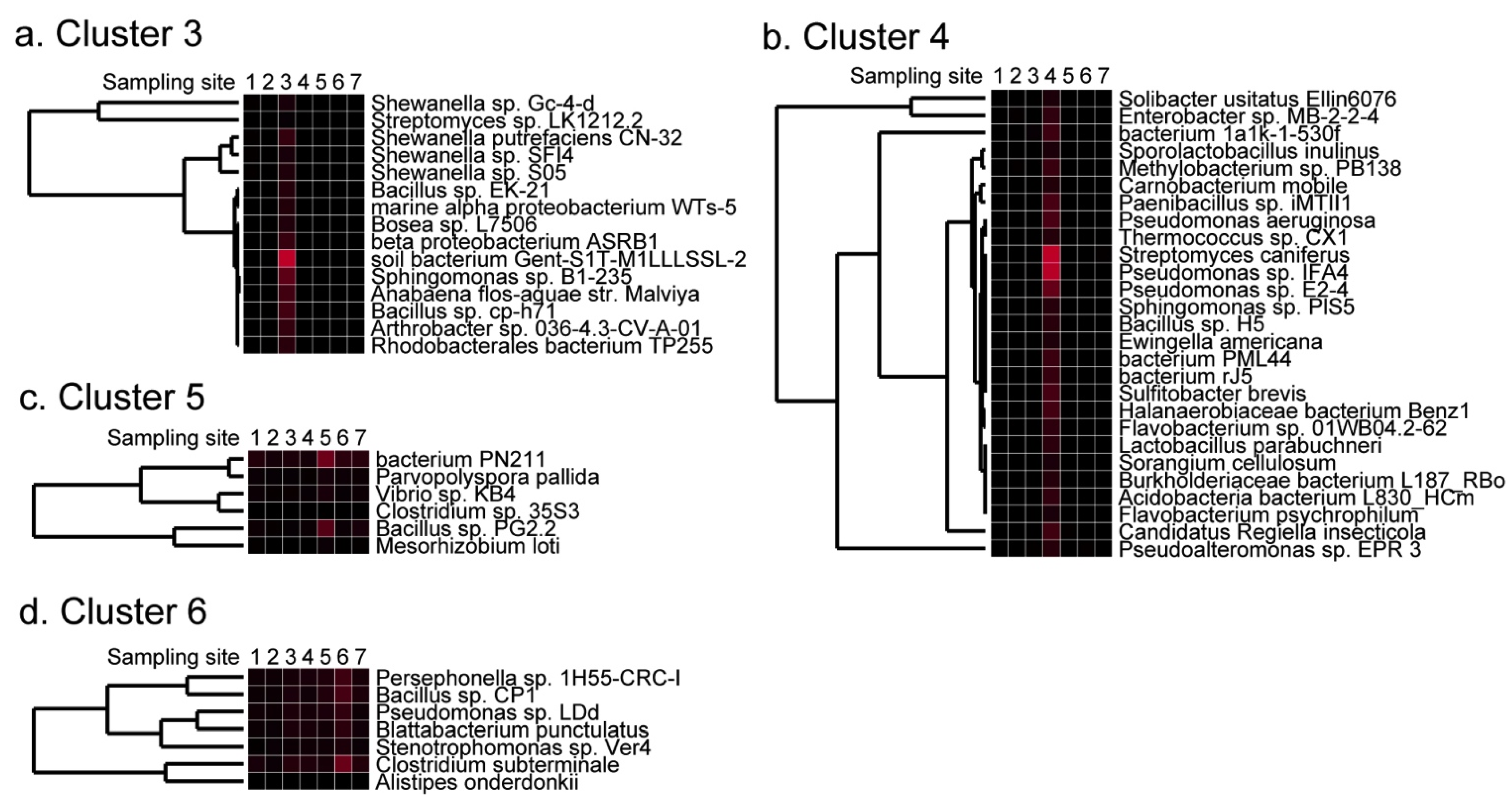
2.4. Detection of Site-Specific Clusters and Species


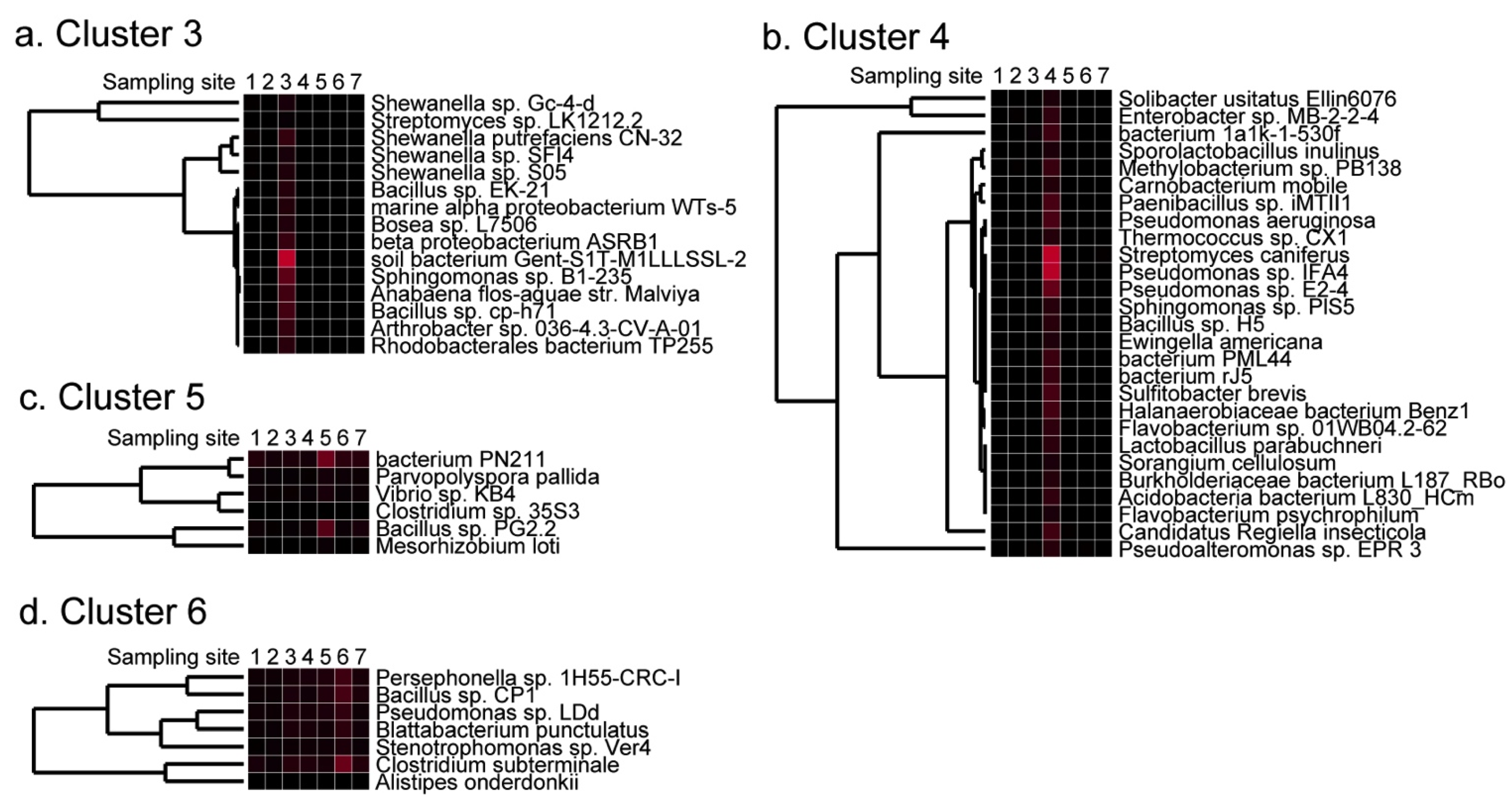
2.5. Discussion of Results
3. Experimental
3.1. DNA Microarray Design
3.2. DNA Extraction from Environmental Water
3.3. Whole Genome Amplification (WGA)
3.4. Fluorescent Labeling, Hybridization and Analysis of DNA Microarray
3.5. Data Analysis of DNA Microarray
3.6. Nested PCR
4. Conclusions
Supplementary Files
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef]
- Amaral-Zettler, L.A.; Mccliment, E.A.; Ducklow, H.W.; Huse, S.M. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS One 2009, 4, e6372. [Google Scholar]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Mark, W.D.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef]
- Rusch, D.B.; Halpern, A.L.; Sutton, G.; Heidelberg, K.B.; Williamson, S.; Yooseph, S.; Wu, D.; Eisen, J.A.; Hoffman, J.M.; Remington, K.; et al. The Sorcerer II Global Ocean Sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007, 5, e77. [Google Scholar] [CrossRef]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L.; et al. Functional metagenomic profiling of nine biomes. Nature 2008, 452, 629–632. [Google Scholar]
- Brodie, E.L.; Desantis, T.Z.; Joyner, D.C.; Baek, S.M.; Larsen, J.T.; Andersen, G.L.; Hazen, T.C.; Richardson, P.M.; Herman, D.J.; Tokunaga, T.K.; et al. Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl. Environ. Microbiol. 2006, 72, 6288–6298. [Google Scholar] [CrossRef]
- Palmer, C.; Bik, E.M.; Eisen, M.B.; Eckburg, P.B.; Sana, T.R.; Wolber, P.K.; Relman, D.A.; Brown, P.O. Rapid quantitative profiling of complex microbial populations. Nucleic Acids Res. 2006, 34, e5. [Google Scholar] [CrossRef]
- Japan Meteorological Agency. Ocean Current. Available online: www.data.kishou.go.jp/db/kaikyo/jun/current_NK.html (accessed on 20 July 2012).
- Japan Meteorological Agency. Monthly Mean Sea Surface Temperature. Available online: www.data.kishou.go.jp/db/kaikyo/monthly/sst.html (accessed on 20 July 2012).
- Wale, N.; Karypis, G. Target fishing for chemical compounds using target-ligand activity data and ranking based methods. J. Chem. Inf. Model 2009, 49, 2190–2201. [Google Scholar] [CrossRef]
- Monfort, P.; Baleux, B. Distribution and survival of motile Aeromonas spp. in brackish water receiving sewage treatment effluent. Appl. Environ. Microbiol. 1991, 57, 2459–2467. [Google Scholar]
- Jolivet-Gougeon, A.; Tamanai-Shacoori, T.; Sauvager, F.; Cormier, M. Production of Escherichia coli group I-like heat-labile enterotoxin by Enterobacteriaceae isolated from environmental water. Microbios 1997, 90, 209–218. [Google Scholar]
- Alexopoulos, A.; Plessas, S.; Voidarou, C.; Noussias, H.; Stavropoulou, E.; Mantzourani, I.; Tzora, A.; Skoufos, I.; Bezirtzoglou, E. Microbial ecology of fish species ongrowing in Greek sea farms and their watery environment. Anaerobe 2011, 17, 264–266. [Google Scholar] [CrossRef]
- Lee, E.S.; Lee, M.Y.; Han, S.H.; Ka, J.O. Occurrence and molecular differentiation of environmental mycobacteria in surface waters. J. Microbiol. Biotechnol. 2008, 18, 1207–1215. [Google Scholar]
- Jernigan, J.A.; Farr, B.M. Incubation period and sources of exposure for cutaneous Mycobacterium marinum infection: Case report and review of the literature. Clin. Infect. Dis. 2000, 31, 439–443. [Google Scholar] [CrossRef]
- Yu, Y.; Li, H.; Zeng, Y.; Chen, B. Extracellular enzymes of cold-adapted bacteria from Arctic sea ice, Canada Basin. Polar Biol. 2009, 32, 1539–1547. [Google Scholar] [CrossRef]
- Takami, H.; Inoue, A.; Fuji, F.; Horikoshi, K. Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiol. Lett. 1997, 152, 279–285. [Google Scholar] [CrossRef]
- Gihring, T.M.; Banfield, J.F. Arsenite oxidation and arsenate respiration by a new Thermus isolate. FEMS Microbiol. Lett. 2001, 204, 335–340. [Google Scholar] [CrossRef]
- Takai, K.; Hirayama, H.; Sakihama, Y.; Inagaki, F.; Yamato, Y.; Horikoshi, K. Isolation and metabolic characteristics of previously uncultured members of the order aquificales in a subsurface gold mine. Appl. Environ. Microbiol. 2002, 68, 3046–3054. [Google Scholar] [CrossRef]
- Hirayama, H.; Takai, K.; Inagaki, F.; Yamato, Y.; Suzuki, M.; Nealson, K.H.; Horikoshi, K. Bacterial community shift along a subsurface geothermal water stream in a Japanese gold mine. Extremophiles 2005, 9, 169–184. [Google Scholar]
- Nakagawa, T.; Ishibashi, J.; Maruyama, A.; Yamanaka, T.; Morimoto, Y.; Kimura, H.; Urabe, T.; Fukui, M. Analysis of dissimilatory sulfite reductase and 16S rRNA gene fragments from deep-sea hydrothermal sites of the Suiyo Seamount, Izu-Bonin Arc, Western Pacific. Appl. Environ. Microbiol. 2004, 70, 393–403. [Google Scholar]
- Kojima, H.; Teske, A.; Fukui, M. Morphological and phylogenetic characterizations of freshwater Thioploca species from Lake Biwa, Japan, and Lake Constance, Germany. Appl. Environ. Microbiol. 2003, 69, 390–398. [Google Scholar] [CrossRef]
- Haramoto, E.; Kitajima, M.; Katayama, H.; Asami, M.; Akiba, M.; Kunikane, S. Application of real-time PCR assays to genotyping of F-specific phages in river water and sediments in Japan. Water Res. 2009, 43, 3759–3764. [Google Scholar] [CrossRef]
- Colwell, R.R. Infectious disease and environment: Cholera as a paradigm for waterborne disease. Int. Microbiol. 2004, 7, 285–289. [Google Scholar]
- Hrudey, S.E.; Hrudey, E.J. Published case studies of waterborne disease outbreaks—Evidence of a recurrent threat. Water Environ. Res. 2007, 79, 233–245. [Google Scholar] [CrossRef]
- Berger, M.; Shiau, R.; Weintraub, J.M. Review of syndromic surveillance: Implications for waterborne disease detection. J. Epidemiol. Community Health 2006, 60, 543–550. [Google Scholar] [CrossRef]
- Edwards, D.D.; Mcfeters, G.A.; Venkatesan, M.I. Distribution of Clostridium perfringens and fecal sterols in a benthic coastal marine environment influenced by the sewage outfall from McMurdo Station, Antarctica. Appl. Environ. Microbiol. 1998, 64, 2596–2600. [Google Scholar]
- Abdelzaher, A.M.; Wright, M.E.; Ortega, C.; Solo-Gabriele, H.M.; Miller, G.; Elmir, S.; Newman, X.; Shih, P.; Bonilla, J.A.; Bonilla, T.D.; et al. Presence of pathogens and indicator microbes at a non-point source subtropical recreational marine beach. Appl. Environ. Microbiol. 2010, 76, 724–732. [Google Scholar] [CrossRef]
- Robben, P.M.; Sibley, L.D. Food- and waterborne pathogens: You are (infected by) what you eat! Microb. Infect. 2004, 6, 406–413. [Google Scholar] [CrossRef]
- Bhan, M.K.; Bahl, R.; Bhatnagar, S. Typhoid and paratyphoid fever. Lancet 2005, 366, 749–762. [Google Scholar] [CrossRef]
- Rich, V.I.; Pham, V.D.; Eppley, J.; Shi, Y.; Delong, E.F. Time-series analyses of Monterey Bay coastal microbial picoplankton using a “genome proxy” microarray. Environ. Microbiol. 2011, 13, 116–134. [Google Scholar] [CrossRef]
- National Institute of Genetics. DNA Data Bank of Japan (DDBJ). Available online: ftp://ftp.ddbj.nig.ac.jp/ddbj_database/16S (accessed on 30 March 2009).
- Tan, J.C.; Patel, J.J.; Tan, A.; Blain, J.C.; Albert, T.J.; Lobo, N.F.; Ferdig, M.T. Optimizing comparative genomic hybridization probes for genotyping and SNP detection in Plasmodium falciparum. Genomics 2009, 93, 543–550. [Google Scholar] [CrossRef]
- Uda, A.; Tanabayashi, K.; Fujita, O.; Hotta, A.; Yamamoto, Y.; Yamada, A. Comparison of whole genome amplification methods for detecting pathogenic bacterial genomic DNA using microarray. Jpn. J. Infect. Dis. 2007, 60, 355–361. [Google Scholar]
- Suzuki, K.; Takigawa, W.; Tanigawa, K.; Nakamura, K.; Ishido, Y.; Kawashima, A.; Wu, H.; Akama, T.; Sue, M.; Yoshihara, A.; et al. Detection of Mycobacterium leprae DNA from archaeological skeletal remains in Japan using whole genome amplification and polymerase chain reaction. PLoS One 2010, 5, e12422. [Google Scholar] [CrossRef]
- Suzuki, M.T.; Giovannoni, S.J. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 1996, 62, 625–630. [Google Scholar]
- Kuroki, H.; Miyamoto, H.; Fukuda, K.; Iihara, H.; Kawamura, Y.; Ogawa, M.; Wang, Y.; Ezaki, T.; Taniguchi, H. Legionella impletisoli sp. nov. and Legionella yabuuchiae sp. nov., isolated from soils contaminated with industrial wastes in Japan. Syst. Appl. Microbiol. 2007, 30, 273–279. [Google Scholar]
- Nubel, U.; Engelen, B.; Felske, A.; Snaidr, J.; Wieshuber, A.; Amann, R.I.; Ludwig, W.; Backhaus, H. Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J. Bacteriol. 1996, 178, 5636–5643. [Google Scholar]
- Akama, T.; Suzuki, K.; Tanigawa, K.; Kawashima, A.; Wu, H.; Nakata, N.; Osana, Y.; Sakakibara, Y.; Ishii, N. Whole-genome tiling array analysis of Mycobacterium leprae RNA reveals high expression of pseudogenes and noncoding regions. J. Bacteriol. 2009, 191, 3321–3327. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, X.; Story, M. Statistical methods of background correction for Illumina BeadArray data. Bioinformatics 2009, 25, 751–757. [Google Scholar] [CrossRef]
- Affymetrix, Inc. Statistical Algorithms Description Document. Technical Report; Affymetrix: Santa Clara, CA, USA, 2002. [Google Scholar]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef]
- Hollibaugh, J.T.; Bano, N.; Ducklow, H.W. Widespread distribution in polar oceans of a 16S rRNA gene sequence with affinity to Nitrosospira-like ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 2002, 68, 1478–1484. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Akama, T.; Kawashima, A.; Tanigawa, K.; Hayashi, M.; Ishido, Y.; Luo, Y.; Hata, A.; Fujitani, N.; Ishii, N.; Suzuki, K. Comprehensive Analysis of Prokaryotes in Environmental Water Using DNA Microarray Analysis and Whole Genome Amplification. Pathogens 2013, 2, 591-605. https://doi.org/10.3390/pathogens2040591
Akama T, Kawashima A, Tanigawa K, Hayashi M, Ishido Y, Luo Y, Hata A, Fujitani N, Ishii N, Suzuki K. Comprehensive Analysis of Prokaryotes in Environmental Water Using DNA Microarray Analysis and Whole Genome Amplification. Pathogens. 2013; 2(4):591-605. https://doi.org/10.3390/pathogens2040591
Chicago/Turabian StyleAkama, Takeshi, Akira Kawashima, Kazunari Tanigawa, Moyuru Hayashi, Yuko Ishido, Yuqian Luo, Akihisa Hata, Noboru Fujitani, Norihisa Ishii, and Koichi Suzuki. 2013. "Comprehensive Analysis of Prokaryotes in Environmental Water Using DNA Microarray Analysis and Whole Genome Amplification" Pathogens 2, no. 4: 591-605. https://doi.org/10.3390/pathogens2040591
APA StyleAkama, T., Kawashima, A., Tanigawa, K., Hayashi, M., Ishido, Y., Luo, Y., Hata, A., Fujitani, N., Ishii, N., & Suzuki, K. (2013). Comprehensive Analysis of Prokaryotes in Environmental Water Using DNA Microarray Analysis and Whole Genome Amplification. Pathogens, 2(4), 591-605. https://doi.org/10.3390/pathogens2040591