



Pathogens 2026, 15(3), 276; https://doi.org/10.3390/pathogens15030276 (registering DOI) - 3 Mar 2026
Abstract
Burkholderia pseudomallei is a saprophytic environmental bacterium and the causative agent of melioidosis, a serious opportunistic infection in tropical regions, including northern Australia. Infection occurs following environmental exposure via percutaneous inoculation, ingestion, or inhalation; however, the environmental reservoirs and transmission pathways responsible for
[...] Read more.
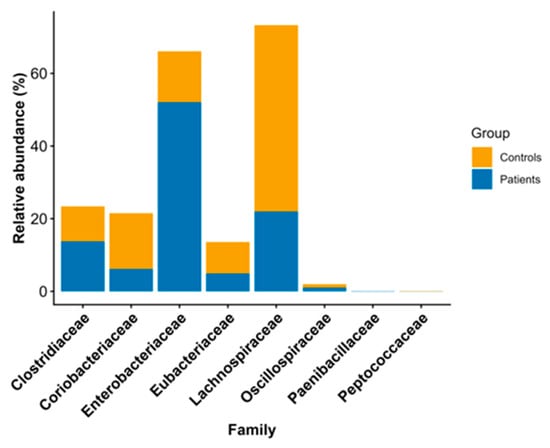
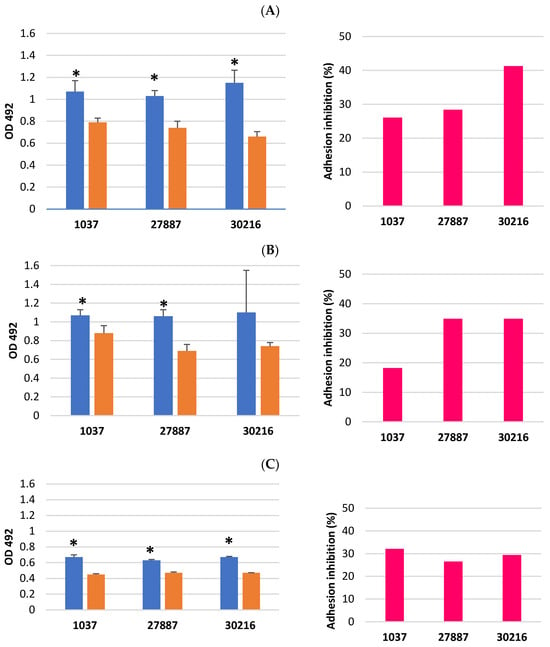
Burkholderia pseudomallei is a saprophytic environmental bacterium and the causative agent of melioidosis, a serious opportunistic infection in tropical regions, including northern Australia. Infection occurs following environmental exposure via percutaneous inoculation, ingestion, or inhalation; however, the environmental reservoirs and transmission pathways responsible for human disease remain poorly defined. Groundwater has been implicated as a potential source of infection, but the factors influencing the persistence and mobility of B. pseudomallei in surface waters in North Queensland are not well understood. Water samples were collected from a groundwater-connected seasonal creek in Townsville, North Queensland, over a 12-month period encompassing wet and dry seasons. Samples were cultured on Ashdown agar and confirmed as B. pseudomallei by qPCR. Multi-locus sequence typing (MLST) was performed using targeted allele sequencing on the Oxford Nanopore MinION platform. Eighteen of 59 water samples were culture-positive for B. pseudomallei. Detection occurred exclusively in turbid, flowing water following ≥30 mm of rainfall and was observed in both wet and dry seasons. MLST of 48 isolates identified 18 sequence types, including 12 novel types. Six sequence types matched previously reported Townsville clinical isolates. These findings indicate that groundwater from a connected urban creek may function as a mobile reservoir for clinically relevant B. pseudomallei strains under specific hydrological and climatic conditions, highlighting rainfall-driven processes as key drivers of environmental exposure risk.
Full article
(This article belongs to the Section Bacterial Pathogens)
►
Show Figures

Figure 1


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}