High-Throughput Sequencing, a VersatileWeapon to Support Genome-Based Diagnosis in Infectious Diseases: Applications to Clinical Bacteriology

Abstract
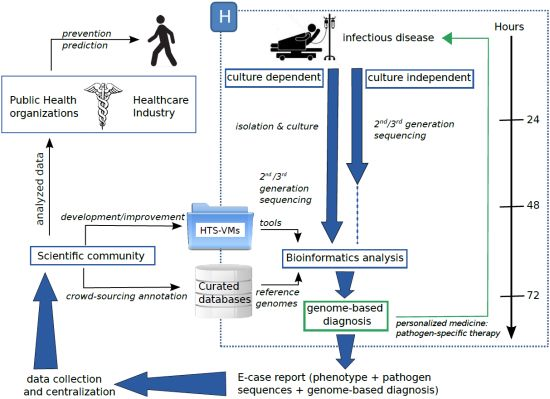
:
1. Introduction
1.1. The Advent of High-Throughput Genomics: A Huge Boost for Pathogens Research

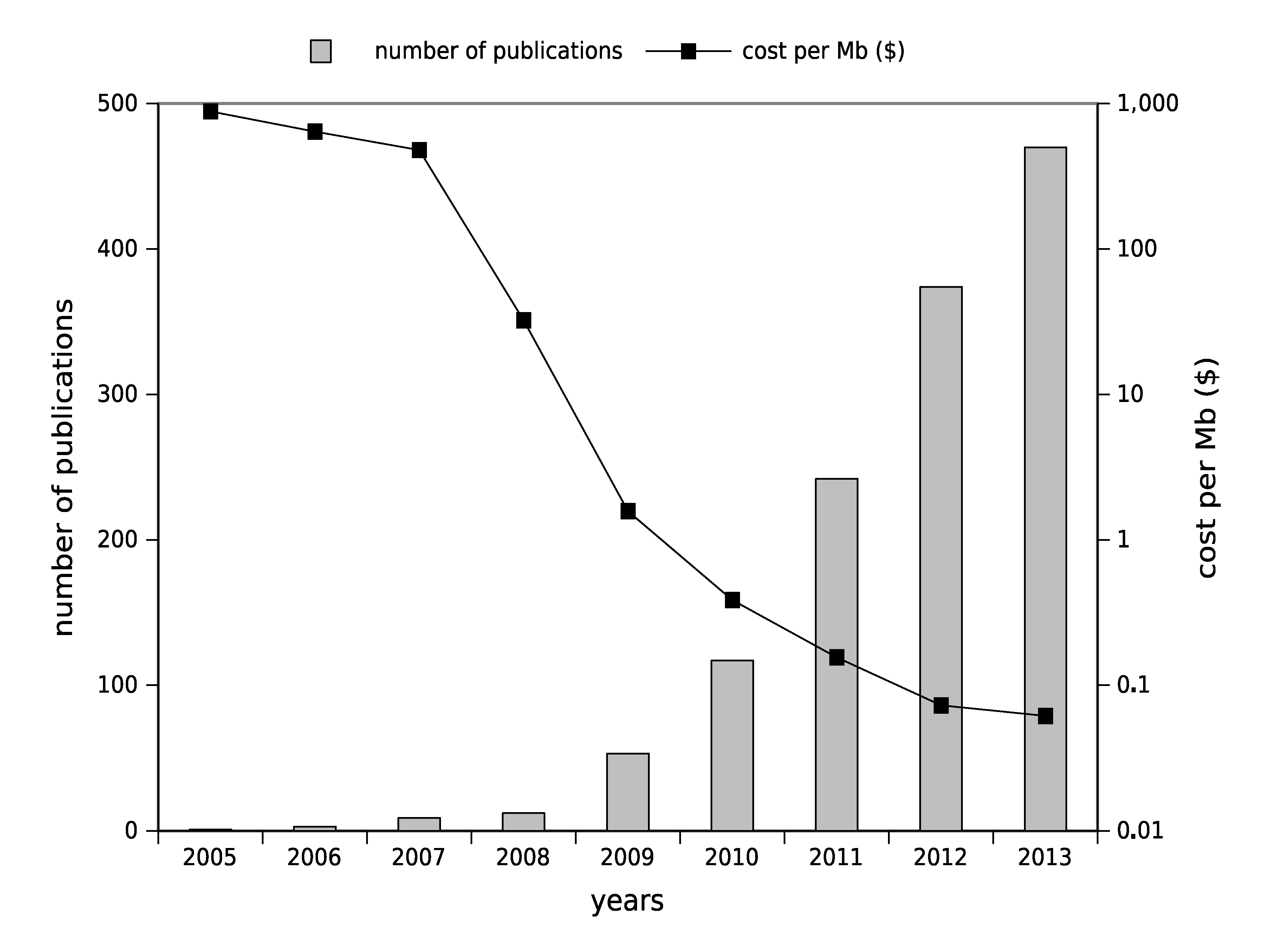
1.2. High-Throughput Sequencing: Toward Routine Use
2. High-Throughput Sequencing toward a New Medicine?
2.1. From PCR-Based Methods for Genotyping to HTS Pilot Studies

{kind=link}
{kind=link}
{kind=link}
| HTS applications | Study highlights | Pathogens/Sample | Real time/retrospective | Platform | Reference | |
|---|---|---|---|---|---|---|
| Culture-dependent | Bacterial genomic epidemiology | feasibility study in a hospital context: improving genetic resolution over common genotyping strategies | S. aureus/clinical samples C. difficile/fecal samples | real time | Illumina MiSeq | [28] |
| pilot study: investigating an outbreak and current limitations for routine use | multidrug-resistant E. coli/rectal swab | retrospective | PGM Ion Torrent | [29] | ||
| WGS data exploring MLST: toward a standardized analysis | C. jejuni and C. coli | retrospective | Illumina HiSeq 2000 | [30] | ||
| WGS to rapidly highlight antibiotic resistance determinants | A. baumannii/tracheal samples | real time | 454-Titanium and Solid version 4 | [31] | ||
| Pathogen evolution | high-resolution genotyping by HTS allowing new insights about an emerging pathogen | methicillin-resistant S. aureus /clinical isolates | retrospective | Illumina GA IIx | [32] | |
| Recombination-filtered core genome to understand pathogen adaptation | E. faecium/isolates from hospitalized patients | retrospective | Illumina GA IIx | [33] | ||
| Culture-independent | Community profiling | proof-of-principle: metagenomics data could be integrated in a diagnosis of cystic fibrosis | airway microbiota in cystic fibrosis/mucolysed sputa | retrospective | PGM Ion Torrent | [34] |
| large-scale study monitoring resistance genes in human gut microbiota | gut microbiota | retrospective | Illumina GA IIx | [35] | ||
| Clinical metagenomics and pathogen discovery | a metagenomics approach to avoid pathogen culture and isolation | Shiga-toxigenic E. coli/stool samples | retrospective | Illumina HiSeq 2500 and MiSeq | [36] | |
| an unbiased method to detect viral pathogens | viral pathogens/nasopharyngeal samples | retrospective | Illumina GA IIx | [37] | ||
| Single-cell microbiology | first evidence of a genome capture from a single cell in a clinical context | P. gingivalis/sink drain | retrospective | Illumina GA IIx | [38] | |
| Immunomagnetic separation for targeted bacterial enrichment with multiple displacement amplification | C. trachomatis/cervical or vaginal swab | retrospective | Illumina GA IIx and HiSeq | [39] | ||
2.2. Bioinformatics to Make Sense of Sequences
| Virus discovery | |||
| Tool | Features | OS | Reference |
| CaPSID | Interactive interface to manage, query and visualize results stored in the database | Linux, Mac | [52] |
| PathSeq | Cloud computingnenvironment | Linux | [53] |
| READSCAN | Genome relativeabundance | Linux, Mac | [54] |
| RINS | Identification of non-human sequences | Linux | [55] |
| VirusFinder | Identification of viruses and integration sites | Linux | [56] |
| Mapping | |||
| Tool | Technology | OS | Reference |
| BFAST | I, So, 4, Hel | Linux, Mac | [57] |
| Bowtie2 | I, 4, Ion | Linux, Mac, Windows | [58] |
| BWA -backtrack | I | Linux | [59] |
| BWA-SW /BWA-MEM | N | Linux | [60] |
| MAQ | I, So | Linux, Mac | [61] |
| Novoalign | I, So, 4, Hel, Ion | Linux | |
| SHRiMP2 | I, So, 4 | Linux, Mac | [62] |
| Smalt | I, 4, Sa, Ion, P | Linux, Mac | |
| Assembly | |||
| Tool | Technology | OS | Reference |
| EULER + Velvet-SC | I | Linux | [63] |
| IDBA -UD | I | Linux | [64] |
| MetaVelvet | I, S, 4 | Linux | [65] |
| MIRA | I, 4, Ion, S | Linux, Mac | [66] |
| Newbler | 4 | Linux | |
| SOAPdenovo | I | Linux | [67] |
| SPAdes | I | Linux, Mac | [68] |
| Velvet | I, S, 4 | Linux, Mac | [69] |
| Sequence annotation | |||
| Tool | Task | OS | Reference |
| BG-7 | Bacterial genome annotation designed for next generation sequencing data | Linux, Mac, Windows | [70] |
| DIYA | Bacterial annotation pipeline | Linux, Mac | [71] |
| PROKKA | Annotation of bacterial, archaeal and viral genomes | Linux, Mac | |
| RAST | Prokaryotic genome annotation service | Linux, Mac, Windows | [72] |
| RATT | Transfer annotation from a reference genome to an unannotated query genome | Linux, Mac | [73] |
3. Current Limitations and Challenges
4. A Possible Future

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Genomes OnLine Database. Available online: http://genomesonline.org (accessed on 27 March 2014).
- Shi, L.; Reid, L.H.; Jones, W.D.; Shippy, R.; Warrington, J.A.; Baker, S.C.; Collins, P.J.; de Longueville, F.; Kawasaki, E.S.; Lee, K.Y.; et al. The MicroArray Quality Control (MAQC) project shows inter-and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006, 24, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Coppee, J.Y. Do DNA microarrays have their future behind them? Microbes Infect. 2008, 10, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Salzberg, S.L.; Pop, M.; Shumway, M.; Umayam, L.; Jiang, L.; Holtzapple, E.; Busch, J.D.; Smith, K.L.; Schupp, J.M.; et al. Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science 2002, 296, 2028–2033. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Constantinidou, C.; Chan, J.Z.; Halachev, M.; Sergeant, M.; Penn, C.W.; Robinson, E.R.; Pallen, M.J. High-throughput bacterial genome sequencing: An embarrassment of choice, a world of opportunity. Nat. Rev. Microbiol. 2012, 10, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Z.; Pallen, M.J.; Oppenheim, B.; Constantinidou, C. Genome sequencing in clinical microbiology. Nat. Biotechnol. 2012, 30, 1068–1071. [Google Scholar] [CrossRef]
- DNA Sequencing Costs. Available online: http://www.genome.gov/sequencingcosts/ (accessed on 27 March 2014).
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Field, D.; Wilson, G.; van der Gast, C. How do we compare hundreds of bacterial genomes? Curr. Opin. Microbiol. 2006, 9, 499–504. [Google Scholar]
- Subramanian, G.; Mural, R.; Hoffman, S.L.; Venter, J.C.; Broder, S. Microbial disease in humans: A genomic perspective. Mol. Diagn. 2001, 6, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hall, N. Advanced sequencing technologies and their wider impact in microbiology. J. Exp. Biol. 2007, 210, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Torok, M.E.; Peacock, S.J. Rapid whole-genome sequencing of bacterial pathogens in the clinical microbiology laboratory–Pipe dream or reality? J. Antimicrob. Chemother. 2012, 67, 2307–2308. [Google Scholar] [CrossRef]
- Andrews-Polymenis, H.L.; Santiviago, C.A.; McClelland, M. Novel genetic tools for studying food-borne Salmonella. Curr. Opin. Biotechnol. 2009, 20, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T. A technological update of molecular diagnostics for infectious diseases. Infect Disord. Drug Targets 2008, 8, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Top, J.; Schouls, L.M.; Bonten, M.J.; Willems, R.J. Multiple-locus variable-number tandem repeat analysis, a novel typing scheme to study the genetic relatedness and epidemiology of Enterococcus faecium isolates. J. Clin. Microbiol. 2004, 42, 4503–4511. [Google Scholar] [CrossRef] [PubMed]
- Diggle, M.A.; Bell, C.M.; Clarke, S.C. Nucleotide sequence-based typing of meningococci directly from clinical samples. J. Med. Microbiol. 2003, 52, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Shariat, N.; Kirchner, M.K.; Sandt, C.H.; Trees, E.; Barrangou, R.; Dudley, E.G. Subtyping of Salmonella enterica serovar newport outbreak isolates by CRISPR-MVLST and determination of the relationship between CRISPR-MVLST and PFGE results. J. Clin. Microbiol. 2013, 51, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Sabat, A.; Gniadkowski, M.; Krzyszton-Russjan, J.; Empel, J.; Miedzobrodzki, J.; Kosowska-Shick, K.; Appelbaum, P.C.; Hryniewicz, W. Comparison of multiple-locus variable-number tandem-repeat analysis with pulsed-field gel electrophoresis, spa typing, and multilocus sequence typing for clonal characterization of Staphylococcus aureus isolates. J. Clin. Microbiol. 2005, 43, 3095–3100. [Google Scholar] [CrossRef]
- Ehrlich, G.D.; Post, J.C. The time is now for gene-and genome-based bacterial diagnostics: “You Say You Want a Revolution”. JAMA Intern. Med. 2013, 173, 1405–1406. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, G.D.; Hu, F.Z.; Shen, K.; Stoodley, P.; Post, J.C. Bacterial plurality as a general mechanism driving persistence in chronic infections. Clin. Orthop. Relat. Res. 2005, 437, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Ruckert, C.; Nubel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rusch-Gerdes, S.; et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef] [PubMed]
- Hiller, N.L.; Ahmed, A.; Powell, E.; Martin, D.P.; Eutsey, R.; Earl, J.; Janto, B.; Boissy, R.J.; Hogg, J.; Barbadora, K.; et al. Generation of genic diversity among Streptococcus pneumoniae strains via horizontal gene transfer during a chronic polyclonal pediatric infection. PLoS Pathog. 2010, 6, e1001108. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.; Loman, N.J.; Bingle, L.; Jumaa, P.; Weinstock, G.M.; Mortiboy, D.; Pallen, M.J. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J. Hosp. Infect. 2010, 75, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Larrat, S.; Poveda, J.D.; Coudret, C.; Fusillier, K.; Magnat, N.; Signori-Schmuck, A.; Thibault, V.; Morand, P. Sequencing assays for failed genotyping with the versant hepatitis C virus genotype assay (LiPA), version 2.0. J. Clin. Microbiol. 2013, 51, 2815–2821. [Google Scholar] [CrossRef]
- Boers, S.A.; van der Reijden, W.A.; Jansen, R. High-throughput multilocus sequence typing: Bringing molecular typing to the next level. PLoS One 2012, 7, e39630. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D.W.; Golubchik, T.; Gordon, N.C.; Bowden, R.; Piazza, P.; Batty, E.M.; Ip, C.L.; Wilson, D.J.; Didelot, X.; O#x2019;Connor, L.; et al. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2012, 2, e001124. [Google Scholar]
- Sherry, N.L.; Porter, J.L.; Seemann, T.; Watkins, A.; Stinear, T.P.; Howden, B.P. Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J. Clin. Microbiol. 2013, 51, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Cody, A.J.; McCarthy, N.D.; Jansen van Rensburg, M.; Isinkaye, T.; Bentley, S.D.; Parkhill, J.; Dingle, K.E.; Bowler, I.C.; Jolley, K.A.; Maiden, M.C. Real-time genomic epidemiological evaluation of human campylobacter isolates by use of whole-genome multilocus sequence typing. J. Clin. Microbiol. 2013, 51, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Rolain, J.M.; Diene, S.M.; Kempf, M.; Gimenez, G.; Robert, C.; Raoult, D. Real-time sequencing to decipher the molecular mechanism of resistance of a clinical pan-drug-resistant Acinetobacter baumannii isolate from Marseille, France. Antimicrob. Agents Chemother. 2013, 57, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.R.; Feil, E.J.; Holden, M.T.; Quail, M.A.; Nickerson, E.K.; Chantratita, N.; Gardete, S.; Tavares, A.; Day, N.; Lindsay, J.A.; et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science 2010, 327, 469–474. [Google Scholar] [CrossRef] [PubMed]
- de Been, M.; van Schaik, W.; Cheng, L.; Corander, J.; Willems, R.J. Recent recombination events in the core genome are associated with adaptive evolution in Enterococcus faecium. Genome Biol. Evol. 2013, 5, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Sengupta, D.J.; Rosenthal, C.; Costa, G.; Spangler, J.; Sims, E.H.; Jacobs, M.A.; Miller, S.I.; Hoogestraat, D.R.; Cookson, B.T.; et al. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PLoS One 2013, 8, e65226. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, X.; Qin, J.; Lu, N.; Cheng, G.; Wu, N.; Pan, Y.; Li, J.; Zhu, L.; Wang, X.; et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun. 2013, 4, 2151. [Google Scholar] [PubMed]
- Loman, N.J.; Constantinidou, C.; Christner, M.; Rohde, H.; Chan, J.Z.; Quick, J.; Weir, J.C.; Quince, C.; Smith, G.P.; Betley, J.R.; et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 2013, 309, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, F.; Ren, L.; Xiong, Z.; Wu, Z.; Dong, J.; Sun, L.; Zhang, T.; Hu, Y.; Du, J.; et al. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J. Clin. Microbiol. 2011, 49, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.S.; Lombardo, M.J.; Ziegler, M.G.; Novotny, M.; Yee-Greenbaum, J.; Badger, J.H.; Tesler, G.; Nurk, S.; Lesin, V.; Brami, D.; et al. Genome of the pathogen Porphyromonas gingivalis recovered from a biofilm in a hospital sink using a high-throughput single-cell genomics platform. Genome Res. 2013, 23, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Seth-Smith, H.M.; Harris, S.R.; Skilton, R.J.; Radebe, F.M.; Golparian, D.; Shipitsyna, E.; Duy, P.T.; Scott, P.; Cutcliffe, L.T.; O’Neill, C.; et al. Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res. 2013, 23, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Koser, C.U.; Holden, M.T.; Ellington, M.J.; Cartwright, E.J.; Brown, N.M.; Ogilvy-Stuart, A.L.; Hsu, L.Y.; Chewapreecha, C.; Croucher, N.J.; Harris, S.R.; et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 2012, 366, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Price, J.R.; Didelot, X.; Crook, D.W.; Llewelyn, M.J.; Paul, J. Whole genome sequencing in the prevention and control of Staphylococcus aureus infection. J. Hosp. Infect. 2013, 83, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Avidor, B.; Girshengorn, S.; Matus, N.; Talio, H.; Achsanov, S.; Zeldis, I.; Fratty, I.S.; Katchman, E.; Brosh-Nissimov, T.; Hassin, D.; et al. Evaluation of a benchtop HIV ultradeep pyrosequencing drug resistance assay in the clinical laboratory. J. Clin. Microbiol. 2013, 51, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Jensen, S.T.; Male, F.; Bittinger, K.; Hodinka, R.L.; Miller, M.D.; Bushman, F.D. Switching between raltegravir resistance pathways analyzed by deep sequencing. AIDS 2011, 25, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Gharizadeh, B.; Norberg, E.; Loffler, J.; Jalal, S.; Tollemar, J.; Einsele, H.; Klingspor, L.; Nyren, P. Identification of medically important fungi by the Pyrosequencing technology. Mycoses 2004, 47, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Stower, H. Pathogen sequencing: Picking and choosing. Nat. Rev. Genet. 2013, 14, 304. [Google Scholar]
- Pallen, M.J.; Loman, N.J.; Penn, C.W. High-throughput sequencing and clinical microbiology: Progress, opportunities and challenges. Curr. Opin. Microbiol. 2010, 13, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Oyola, S.O.; Gu, Y.; Manske, M.; Otto, T.D.; O’Brien, J.; Alcock, D.; Macinnis, B.; Berriman, M.; Newbold, C.I.; Kwiatkowski, D.P.; et al. Efficient depletion of host DNA contamination in malaria clinical sequencing. J. Clin. Microbiol. 2013, 51, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Sboner, A.; Mu, X.J.; Greenbaum, D.; Auerbach, R.K.; Gerstein, M.B. The real cost of sequencing: higher than you think. Genome Biol. 2011, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Software list, Available online: http://seqanswers.com/wiki/Software/list (accessed on 27 March 2014).
- Chiu, C.Y. Viral pathogen discovery. Curr. Opin. Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, N.A.; Rung, J.; Brazma, A.; Marioni, J.C. Tools for mapping high-throughput sequencing data. Bioinformatics 2012, 28, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Borozan, I.; Wilson, S.; Blanchette, P.; Laflamme, P.; Watt, S.N.; Krzyzanowski, P.M.; Sircoulomb, F.; Rottapel, R.; Branton, P.E.; Ferretti, V. CaPSID: A bioinformatics platform for computational pathogen sequence identification in human genomes and transcriptomes. BMC Bioinf. 2012, 13, 206. [Google Scholar] [CrossRef]
- Kostic, A.D.; Ojesina, A.I.; Pedamallu, C.S.; Jung, J.; Verhaak, R.G.; Getz, G.; Meyerson, M. PathSeq: Software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol. 2011, 29, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Naeem, R.; Rashid, M.; Pain, A. READSCAN: a fast and scalable pathogen discovery program with accurate genome relative abundance estimation. Bioinformatics 2013, 29, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Qu, K.; Lee, C.S.; Ungewickell, A.; Khavari, P.A. Rapid identification of non-human sequences in high-throughput sequencing datasets. Bioinformatics 2012, 28, 1174–1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, P.; Zhao, Z. VirusFinder: Software for efficient and accurate detection of viruses and their integration sites in host genomes through next generation sequencing data. PLoS One 2013, 8, e64465. [Google Scholar] [CrossRef] [PubMed]
- Homer, N.; Merriman, B.; Nelson, S.F. BFAST: An alignment tool for large scale genome resequencing. PLoS One 2009, 4, e7767. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Dzamba, M.; Lister, D.; Ilie, L.; Brudno, M. SHRiMP2: Sensitive yet practical short read mapping. Bioinformatics 2011, 27, 1011–1012. [Google Scholar] [CrossRef] [PubMed]
- Chitsaz, H.; Yee-Greenbaum, J.L.; Tesler, G.; Lombardo, M.J.; Dupont, C.L.; Badger, J.H.; Novotny, M.; Rusch, D.B.; Fraser, L.J.; Gormley, N.A.; et al. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat. Biotechnol. 2011, 29, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Namiki, T.; Hachiya, T.; Tanaka, H.; Sakakibara, Y. MetaVelvet: An extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012, 40, e155. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using trace signals and additional sequence information. In Proceedings of the Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB), Hannover, Germany , 1999; pp. 45–56.
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Pareja-Tobes, P.; Manrique, M.; Pareja-Tobes, E.; Pareja, E.; Tobes, R. BG7: A new approach for bacterial genome annotation designed for next generation sequencing data. PLoS One 2012, 7, e49239. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.C.; Osborne, B.; Read, T.D. DIYA: A bacterial annotation pipeline for any genomics lab. Bioinformatics 2009, 25, 962–963. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; deJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.D.; Dillon, G.P.; Degrave, W.S.; Berriman, M. RATT: Rapid annotation transfer tool. Nucleic Acids Res. 2011, 39, e57. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.J.; Wattam, A.R.; Cammer, S.A.; Gabbard, J.L.; Shukla, M.P.; Dalay, O.; Driscoll, T.; Hix, D.; Mane, S.P.; Mao, C.; et al. PATRIC: The comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 2011, 79, 4286–4298. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Pop, M. ARDB–Antibiotic resistance genes database. Nucleic Acids Res. 2009, 37, D443–D447. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–352. [Google Scholar] [CrossRef] [PubMed]
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinf. 2013, 15, 256–278. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar.; Buchner, A.; Lai, T.; Steppi, S.; Jobb, G.; Frster, W.; et al. ARB: A software environment for sequence data. Nucleic Acids Research 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.C. The missing graphical user interface for genomics. Genome Biol. 2010, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Afgan, E.; Ananda, G.; Baker, D.; Blankenberg, D.; Chakrabarty, R.; Coraor, N.; Goecks, J.; et al. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef] [PubMed]
- Krampis, K.; Booth, T.; Chapman, B.; Tiwari, B.; Bicak, M.; Field, D.; Nelson, K.E. Cloud BioLinux: Pre-configured and on-demand bioinformatics computing for the genomics community. BMC Bioinformatics 2012, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.; Lee, H.; Maurer-Stroh, S.; Molnar, M.; Pongor, S.; Eisenhaber, B.; Eisenhaber, F. How bioinformatics influences health informatics: Usage of biomolecular sequences, expression profiles and automated microscopic image analyses for clinical needs and public health. Health Inf. Sci. Syst. 2013, 1, 2. [Google Scholar] [CrossRef]
- Frank, C.; Faber, M.S.; Askar, M.; Bernard, H.; Fruth, A.; Gilsdorf, A.; Hohle, M.; Karch, H.; Krause, G.; Prager, R.; et al. Large and ongoing outbreak of haemolytic uraemic syndrome, Germany, May 2011. Euro Surveill. 2011, 16. [Google Scholar]
- Gullapalli, R.R.; Desai, K.V.; Santana-Santos, L.; Kant, J.A.; Becich, M.J. Next generation sequencing in clinical medicine: Challenges and lessons for pathology and biomedical informatics. J. Pathol. Inform. 2012, 3, 40. [Google Scholar] [PubMed]
- Najafzadeh, M.; Lynd, L.D.; Davis, J.C.; Bryan, S.; Anis, A.; Marra, M.; Marra, C.A. Barriers to integrating personalized medicine into clinical practice: a best-worst scaling choice experiment. Genet. Med. 2012, 14, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Lampa, S.; Dahlo, M.; Olason, P.I.; Hagberg, J.; Spjuth, O. Lessons learned from implementing a national infrastructure in Sweden for storage and analysis of next-generation sequencing data. Gigascience 2013, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Carrico, J.A.; Sabat, A.J.; Friedrich, A.W.; Ramirez, M. Bioinformatics in bacterial molecular epidemiology and public health: Databases, tools and the next-generation sequencing revolution. Euro Surveill. 2013, 18, 20382. [Google Scholar]
- Hong, H.; Zhang, W.; Shen, J.; Su, Z.; Ning, B.; Han, T.; Perkins, R.; Shi, L.; Tong, W. Critical role of bioinformatics in translating huge amounts of next-generation sequencing data into personalized medicine. Sci. China Life Sci. 2013, 56, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Dunne, W.M.; Westblade, L.F.; Ford, B. Next-generation and whole-genome sequencing in the diagnostic clinical microbiology laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Nocq, J.; Celton, M.; Gendron, P.; Lemieux, S.; Wilhelm, B.T. Harnessing virtual machines to simplify next-generation DNA sequencing analysis. Bioinformatics 2013, 29, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Taylor, J. Next-generation sequencing data interpretation: Enhancing reproducibility and accessibility. Nat. Rev. Genet. 2012, 13, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A.; Altshuler, D.; Durbin, R.M.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, F.J.; Heymann, M.; Ternes, A.M.; Wienecke-Baldacchino, A.; Struck, D.; Moes, D.; Schneider, R. Bioinformatics as a driver, not a passenger, of translational biomedical research: Perspectives from the 6th Benelux bioinformatics conference. J. Clin. Bioinforma 2012, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, H.; Sarkans, U.; Kolesnikov, N.; Abeygunawardena, N.; Burdett, T.; Dylag, M.; Emam, I.; Farne, A.; Hastings, E.; Holloway, E.; et al. ArrayExpress update–an archive of microarray and high-throughput sequencing-based functional genomics experiments. Nucleic Acids Res. 2011, 39, D1002–D1004. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea, J.A. Infection systems biology: From reactive to proactive (P4) medicine. Int. Microbiol. 2012, 15, 55–60. [Google Scholar] [PubMed]
- Collins, F.S.; Hamburg, M.A. First FDA authorization for next-generation sequencer. N. Engl. J. Med. 2013, 369, 2369–2371. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Caboche, S.; Audebert, C.; Hot, D. High-Throughput Sequencing, a VersatileWeapon to Support Genome-Based Diagnosis in Infectious Diseases: Applications to Clinical Bacteriology. Pathogens 2014, 3, 258-279. https://doi.org/10.3390/pathogens3020258
Caboche S, Audebert C, Hot D. High-Throughput Sequencing, a VersatileWeapon to Support Genome-Based Diagnosis in Infectious Diseases: Applications to Clinical Bacteriology. Pathogens. 2014; 3(2):258-279. https://doi.org/10.3390/pathogens3020258
Chicago/Turabian StyleCaboche, Ségolène, Christophe Audebert, and David Hot. 2014. "High-Throughput Sequencing, a VersatileWeapon to Support Genome-Based Diagnosis in Infectious Diseases: Applications to Clinical Bacteriology" Pathogens 3, no. 2: 258-279. https://doi.org/10.3390/pathogens3020258
APA StyleCaboche, S., Audebert, C., & Hot, D. (2014). High-Throughput Sequencing, a VersatileWeapon to Support Genome-Based Diagnosis in Infectious Diseases: Applications to Clinical Bacteriology. Pathogens, 3(2), 258-279. https://doi.org/10.3390/pathogens3020258