
Molecular Epidemiology and Spatio-Temporal Dynamics of the H3N8 Equine Influenza Virus in South America

 and
and
Abstract
:1. Introduction
2. Results
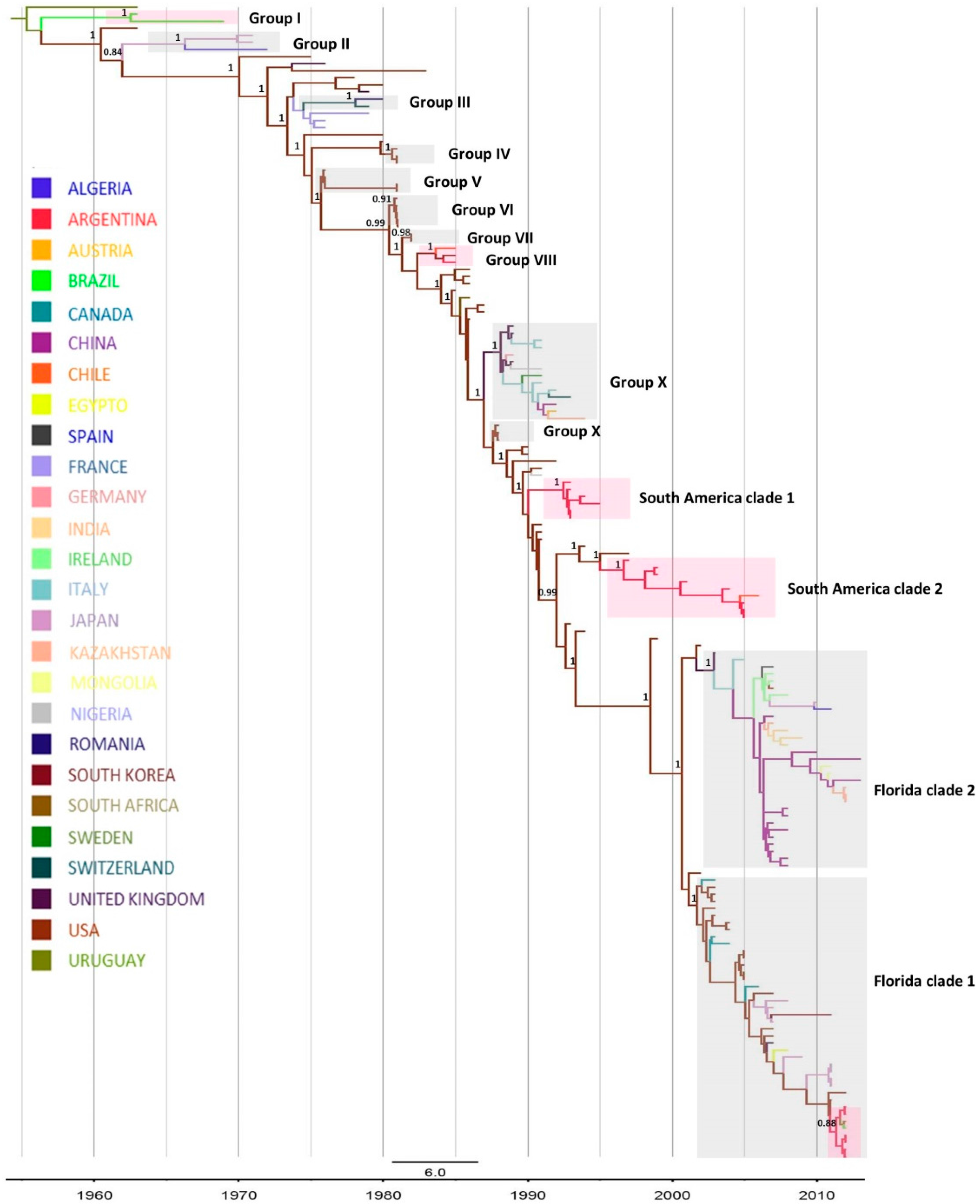
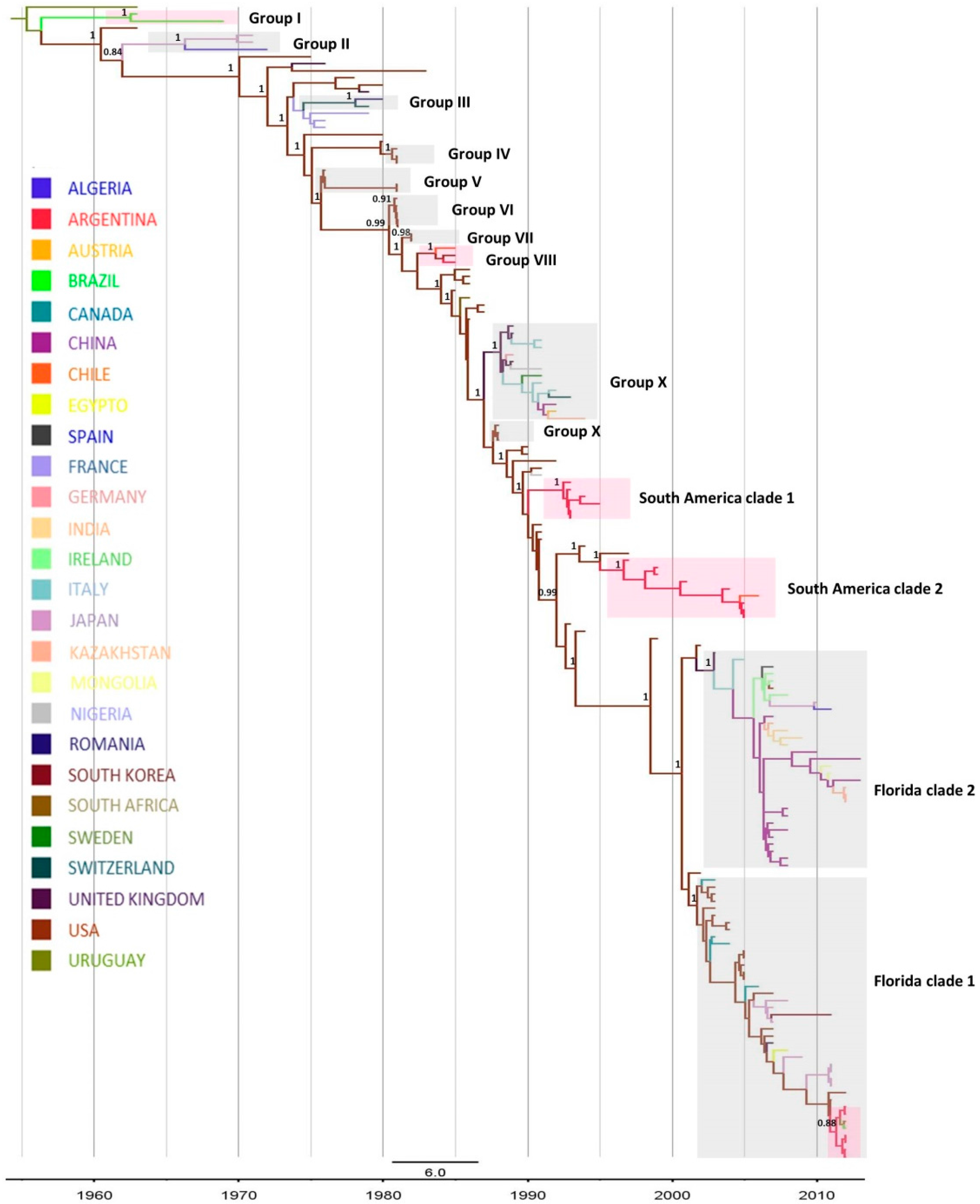
2.1. Phylogenetic Analysis
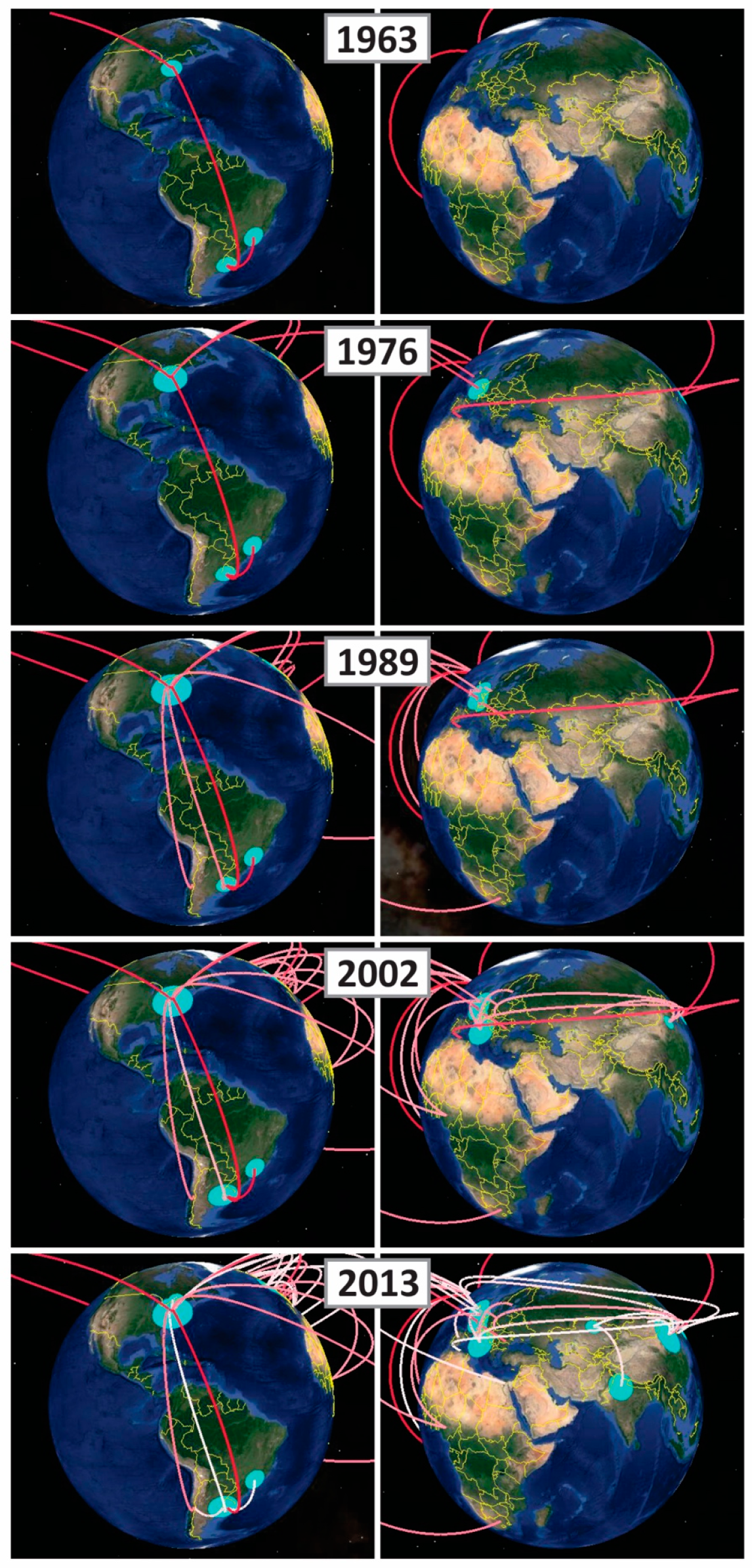
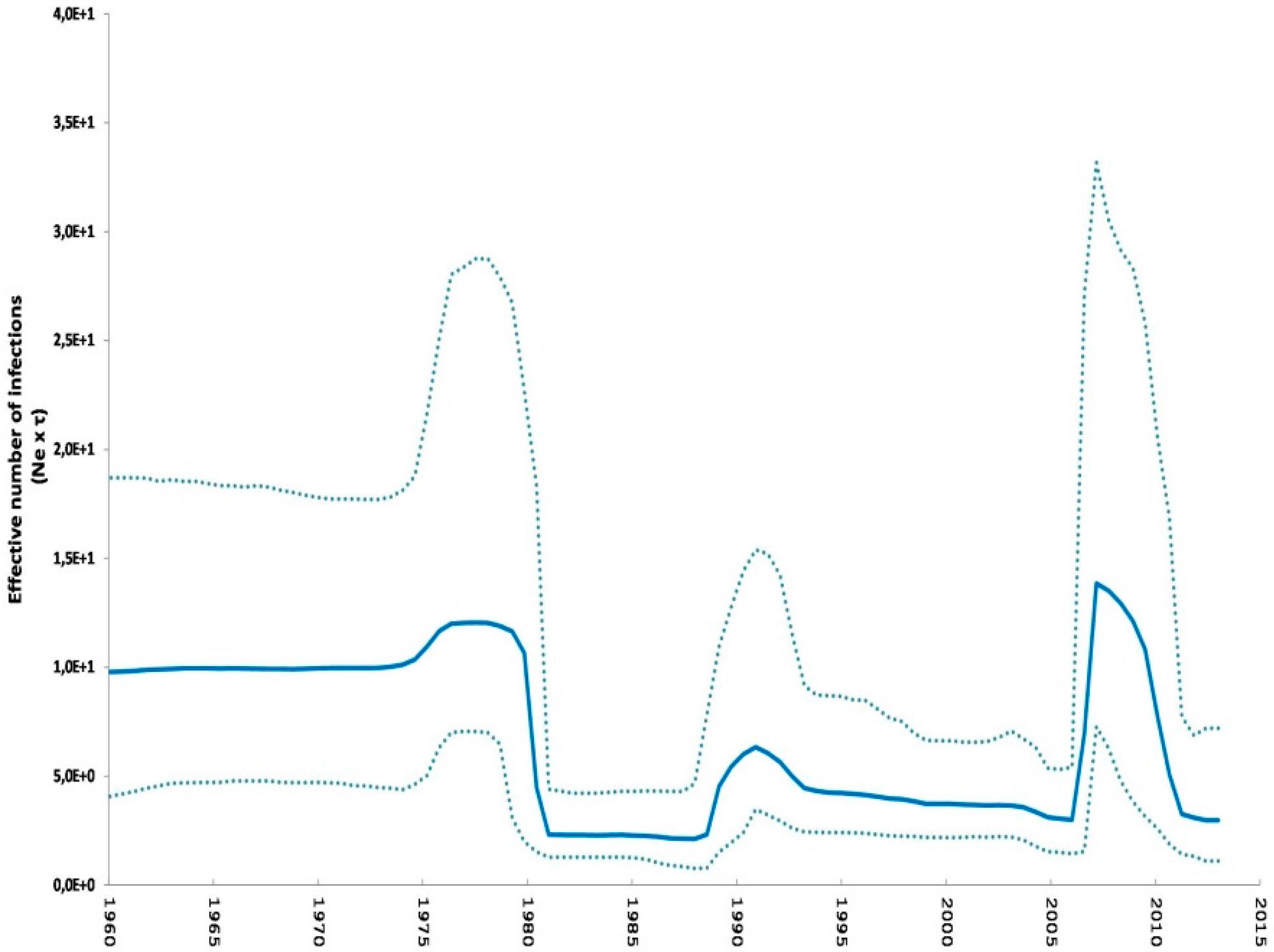
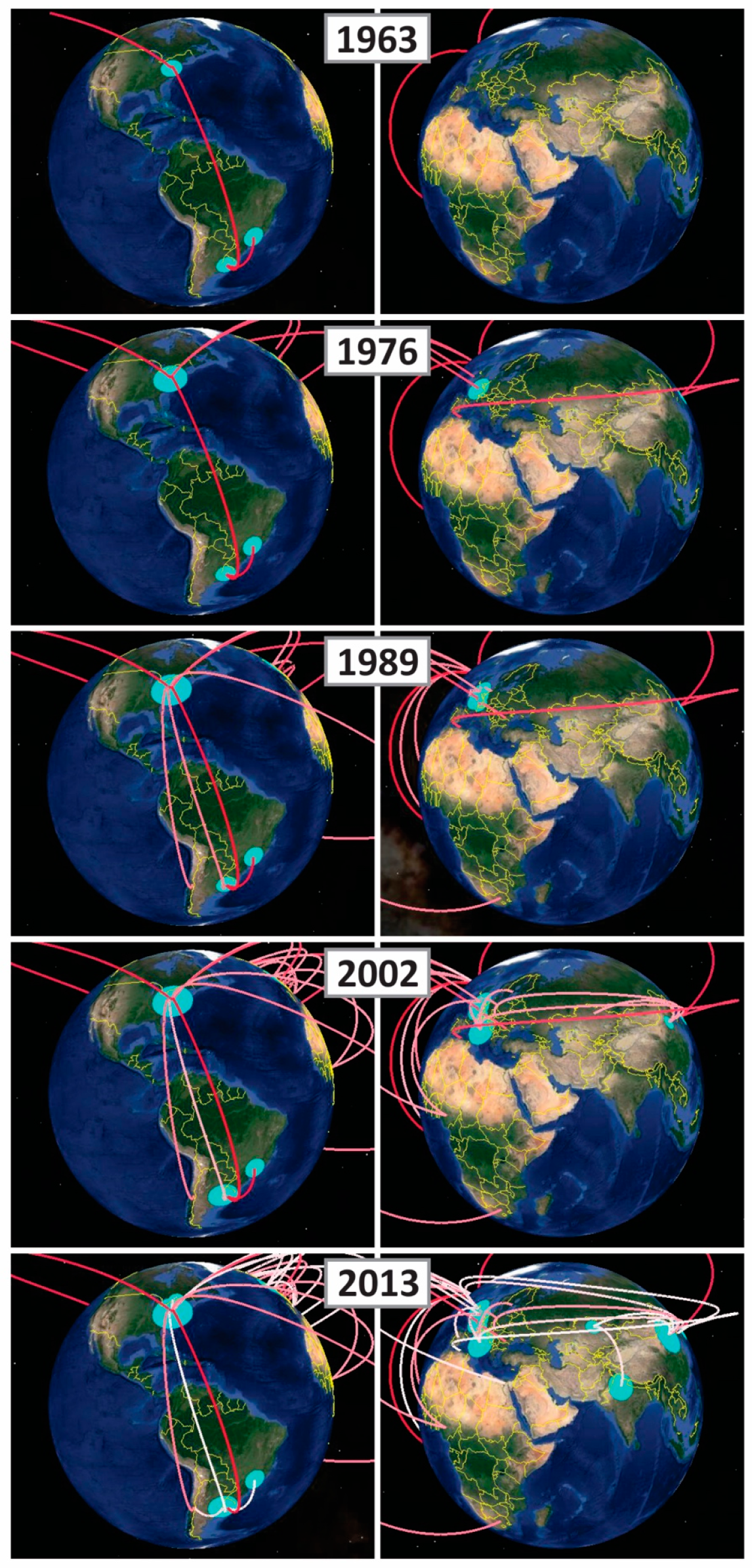
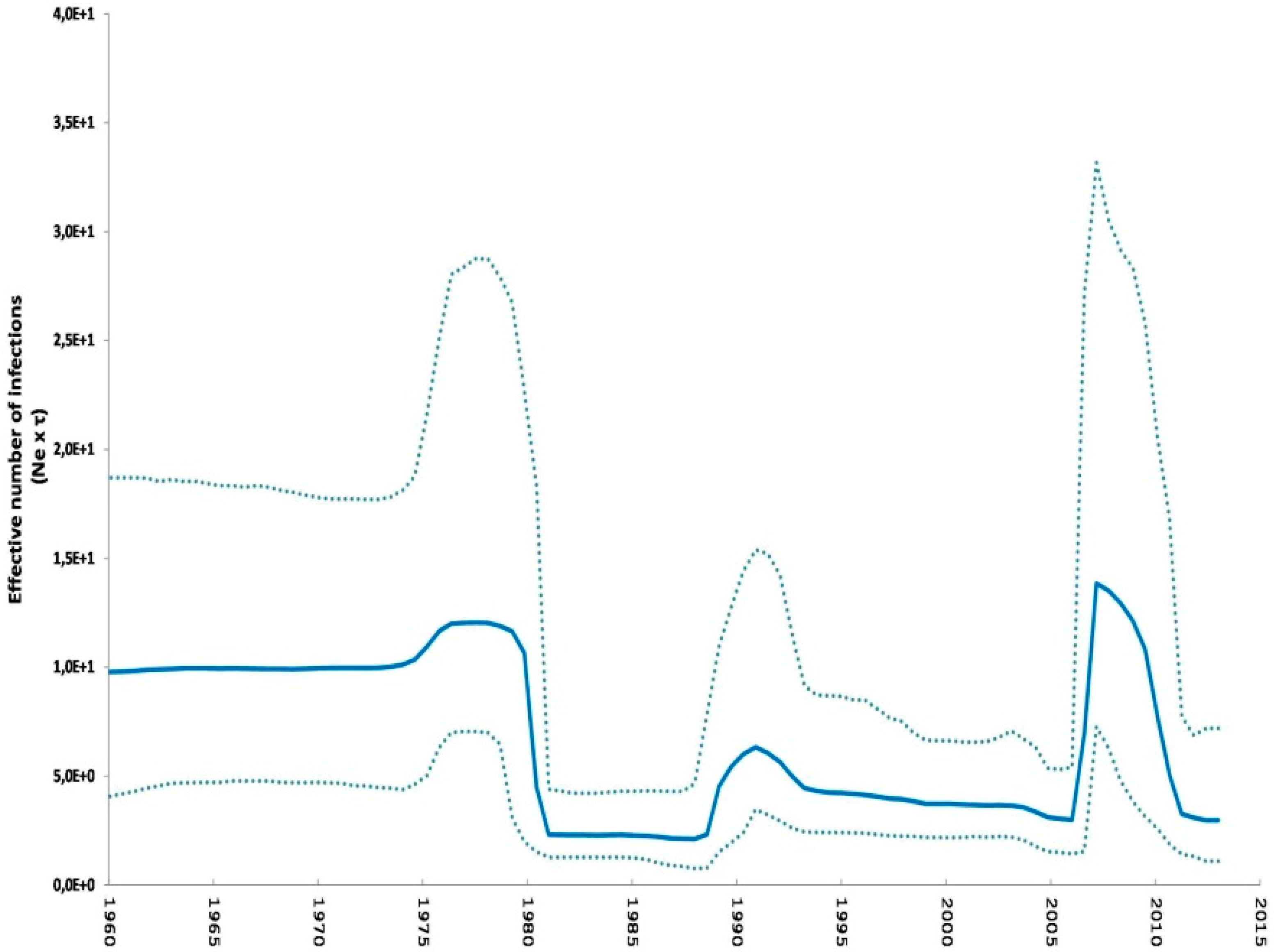
2.2. Phylodynamic Analysis
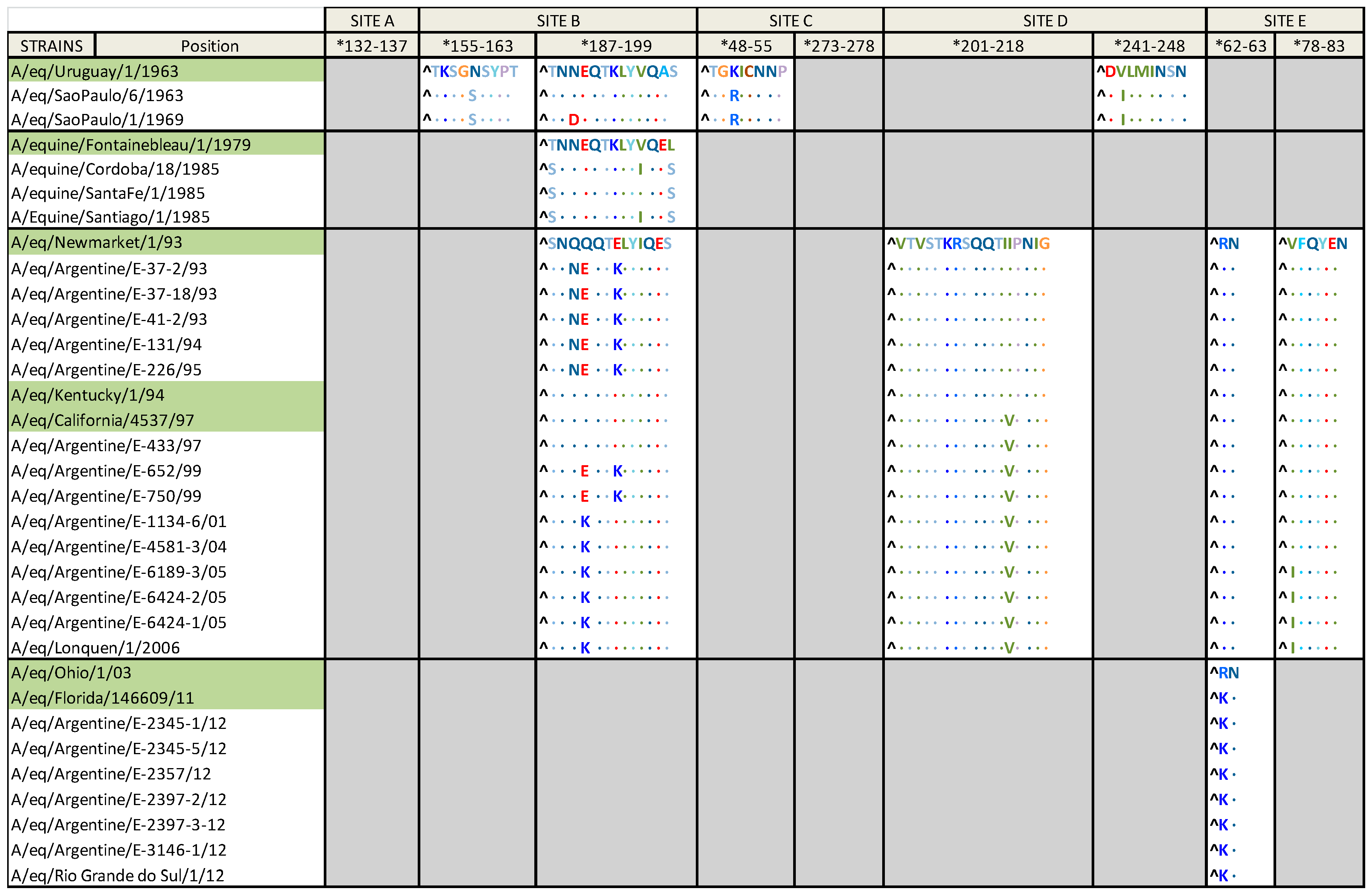
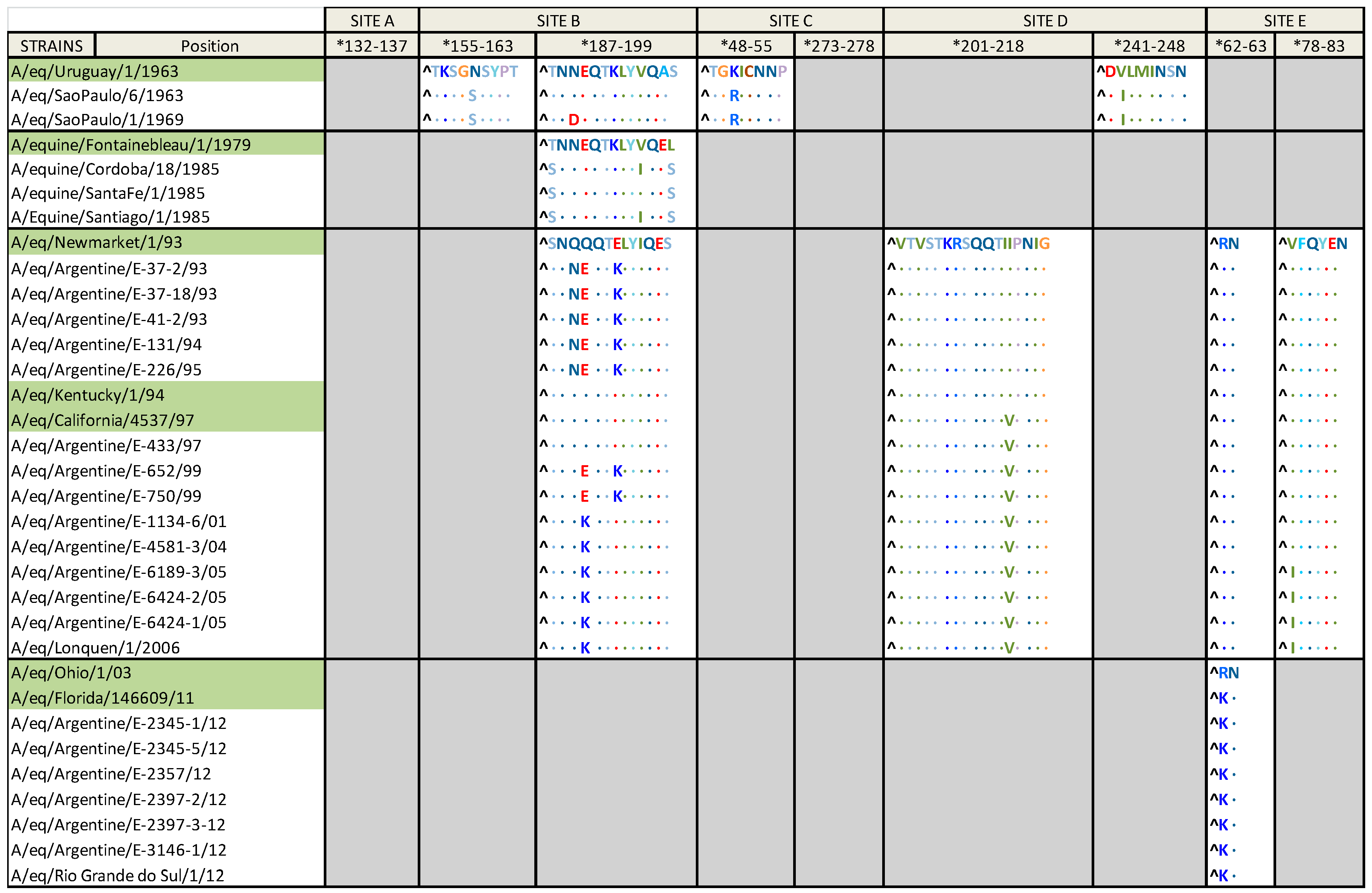
2.3. Amino Acid Alignment
3. Discussion
4. Experimental Section
4.1. HA Gene Sequencing
4.2. Sequence Analysis and Phylogenetic Trees
4.3. Phylodynamic Analysis
4.4. Amino Acid Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Woodward, A.L.; Rash, A.S.; Blinman, D.; Bowman, S.; Chambers, T.M.; Daly, J.M.; Damiani, A.; Joseph, S.; Lewis, N.; McCauley, J.W.; et al. Development of a surveillance scheme for equine influenza in the UK and characterisation of viruses isolated in Europe, Dubai and the USA from 2010–2012. Vet. Microbiol. 2014, 169, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Gildea, S.; Arkins, S.; Cullinane, A. Management and environmental factors involved in equine influenza outbreaks in Ireland 2007–2010. Equine Vet. J. 2011, 43, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, A.; Elton, D.; Mumford, J. Equine influenza—Surveillance and control. Influenza Other Respir. Viruses 2010, 4, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Sovinova, O.; Tumova, B.; Pouska, F.; Nemec, J. Isolation of a virus causing respiratory disease in horses. Acta Virol. 1958, 2, 52–61. [Google Scholar] [PubMed]
- Scholtens, R.G.; Steele, J.H.; Dowdle, W.R.; Yarbrough, W.B.; Robinson, R.Q. U.S. Epizootic of equine influenza, 1963. Public Health Rep. 1964, 79, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Waddell, G.H.; Teigland, M.B.; Sigel, M.M. A new influenza virus associated with equine respiratory disease. J. Am. Vet. Med. Assoc. 1963, 143, 587–590. [Google Scholar]
- Webster, R.G. Are equine 1 influenza viruses still present in horses? Equine Vet. J. 1993, 25, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, Y.; Bean, W.J.; Webster, R.G. Evolution of the hemagglutinin of equine h3 influenza viruses. Virology 1989, 169, 283–292. [Google Scholar] [CrossRef]
- Daly, J.M.; MacRae, S.; Newton, J.R.; Wattrang, E.; Elton, D.M. Equine influenza: A review of an unpredictable virus. Vet. J. 2011, 189, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.M.; Lai, A.C.; Binns, M.M.; Chambers, T.M.; Barrandeguy, M.; Mumford, J.A. Antigenic and genetic evolution of equine H3N8 influenza a viruses. J. Gen. Virol. 1996, 77, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Chambers, T.M.; Holland, R.E., Jr.; Morley, P.S.; Haines, D.M.; Townsend, H.G.; Barrandeguy, M. Diverged evolution of recent equine-2 influenza (H3N8) viruses in the western hemisphere. Arch. Virol. 2001, 146, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Gildea, S.; Quinlivan, M.; Arkins, S.; Cullinane, A. The molecular epidemiology of equine influenza in ireland from 2007–2010 and its international significance. Equine Vet. J. 2012, 44, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Gildea, S.; Fitzpatrick, D.A.; Cullinane, A. Epidemiological and virological investigations of equine influenza outbreaks in ireland (2010–2012). Influenza Other Respir. Viruses 2013, 7 (Suppl. 4), 61–72. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Nagai, M.; Hayakawa, Y.; Komae, H.; Murakami, N.; Yotsuya, S.; Asakura, S.; Sakoda, Y.; Kida, H. Genetic analyses of an H3N8 influenza virus isolate, causative strain of the outbreak of equine influenza at the Kanazawa racecourse in Japan in 2007. Vet. Med. Sci. 2008, 70, 899–906. [Google Scholar] [CrossRef]
- King, E.; Macdonal, D. Report of the board of inquiry appointed by the board of the national horseracing authority to conduct enquiry into the causes of the equine influenza which started in the western cape in early december 2003 and spread to the eastern cape and gauteng. Aust. Vet. J. 2004, 23, 139–142. [Google Scholar]
- Perglione, C.O.; Gildea, S.; Rimondi, A.; Mino, S.; Vissani, A.; Carossino, M.; Cullinane, A.; Barrandeguy, M. Epidemiological and virological findings during multiple outbreaks of equine influenza in south america in 2012. Influenza Other Respir. Viruses 2016, 10, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.; Daniels, P.; Kirkland, P.; Carroll, A.; Jeggo, M. The 2007 outbreak of equine influenza in Australia: Lessons learned for international trade in horses. Rev. Sci. Tech. 2011, 30, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Yondon, M.; Heil, G.L.; Burks, J.P.; Zayat, B.; Waltzek, T.B.; Jamiyan, B.O.; McKenzie, P.P.; Krueger, W.S.; Friary, J.A.; Gray, G.C. Isolation and characterization of H3N8 equine influenza a virus associated with the 2011 epizootic in mongolia. Influenza Other Respir. Viruses 2013, 7, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Virmani, N.; Bera, B.C.; Singh, B.K.; Shanmugasundaram, K.; Gulati, B.R.; Barua, S.; Vaid, R.K.; Gupta, A.K.; Singh, R.K. Equine influenza outbreak in India (2008-09): Virus isolation, sero-epidemiology and phylogenetic analysis of ha gene. Vet. Microbiol. 2010, 143, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Guo, W.; Huang, W.Q.; Li, H.M.; Zhao, L.P.; Dai, L.L.; He, N.; Hao, X.F.; Xiang, W.H. Genetic evolution of equine influenza viruses isolated in China. Arch. Virol. 2010, 155, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Murcia, P.R.; Wood, J.L.; Holmes, E.C. Genome-scale evolution and phylodynamics of equine H3N8 influenza a virus. J. Virol. 2011, 85, 5312–5322. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.; Pinto, E.; Santibanez, M.C.; Celedon, M.O.; Valenzuela, P.D. Isolation and characterization of the equine influenza virus causing the 2006 outbreak in Chile. Vet. Microbiol. 2009, 137, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Fuschlocher, F.; Zurita, L.; Latorre, G.; Palavicino, I. Influenza equina en la provincia de santiago. In V Convención Nacional de Medicina Veterinaria; Boletin: Valdivia, Chile, 1963; pp. 40–46. [Google Scholar]
- Casanova, A.; Martínez, I.; Román, M. Aislamiento y tipificación del virus de influenza equina en Chile. Arch. Med. Vet. 1977, 9, 91–93. [Google Scholar]
- Berríos, P. Influenza equina en chile (1963–1992). Un posible caso en un ser humano. Rev. Chil. Infect. 2005, 22, 47–50. [Google Scholar] [CrossRef]
- Celedón, M.O.; De Negri, L.; Santibáñez, M.; Berríos, P. Brote de influenza equina en Chile causado por el subtipo H3N8. Agro-Ciencia 1992, 8, 47–48. [Google Scholar]
- OIE: World Animal Health Information Database (WAHID) Interface. Available online: http://www.oie.int/wahis_2/public/wahid.php/Wahidhome/Home (accessed on 26 October 2016).
- Mancini, D.A.; Mendonca, R.M.; Pinto, J.R.; Mori, E.; Fernandes, W. Anti-human influenza protector antibody detected in horses as a zoonotic viruses. Braz. J. Vet. Res. Anim. Sci. 2004, 41, 379–383. [Google Scholar] [CrossRef]
- Alves Beuttemmuller, E.; Woodward, A.; Rash, A.; Dos Santos Ferraz, L.E.; Fernandes Alfieri, A.; Alfieri, A.A.; Elton, D. Characterisation of the epidemic strain of H3N8 equine influenza virus responsible for outbreaks in South America in 2012. Virol. J. 2016, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Fain Binda, J.; Martin, J. Aislamiento del agente etiológico de la epizootia de influenza equina en rosario (Argentina) 1976: Myxovirus influenza a/equi 1/rosario 76. Revista de Medicina Veterinaria Buenos Aires 1977, 58, 261–268. [Google Scholar]
- Nosetto, E.; Pecoraro, M.; Galosi, C.m.; Massone, R.; Cid de la Paz, V.; Ando, R.; Ando, Y.; Etcheverrigaray, M.E. Isolation of an equine influenza virus strain and epizootiological study of the 1985–86 outbreak in Argentina. Rev. Sci. Tech. Off. Int. Epizoot. 1989, 8, 123–128. [Google Scholar] [CrossRef]
- Barrandeguy, M.; Bryans, J.; San Roman, A.; Parreño, V.; Ricci, L.; Schudel, A.; Kness, V.; Fernandez, F. Hallazgos virológicos y serológicos en un brote de influenza equina en los hipódromos de palermo y san isidro ocurrido en abril de 1993. In Proceedings of the XI Jornadas de Actualización Técnico Científicas de la Asociación Argentina de Veterinaria Equina, La Plata, Buenos Aires, Argentina, 9–19 December 1993.
- Miño, S.; Vissani, A.; Trono, K.; Barrandeguy, M. Evolución genética de virus de influenza equina-2 (H3N8) detectados en argentina en los últimos años. In Proceedings of the Congreso Panamericano de Ciencias Veterinarias, Santiago, Chile, 13–16 November 2006.
- Lewis, N.S.; Daly, J.M.; Russell, C.A.; Horton, D.L.; Skepner, E.; Bryant, N.A.; Burke, D.F.; Rash, A.S.; Wood, J.L.; Chambers, T.M.; et al. Antigenic and genetic evolution of equine influenza a (H3N8) virus from 1968 to 2007. J. Virol. 2011, 85, 12742–12749. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Ann. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, A.; Newton, J.R. Equine influenza—A global perspective. Vet. Microbiol. 2013, 167, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.; Cullinane, A. Equine influenza: Antigenic drift and implications for vaccines. Equine Vet. J. 2013, 45, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Olguin Perglione, C.; Golemba, M.D.; Barrandeguy, M. Molecular evolution of H3N8 equine influenza virus in argentina. In Proceedings of the X International Conferences on Equine Infecious Diseases, Buenos Aires, Argentina, 4–8 April 2016; p. S74.
- Worobey, M.; Han, G.Z.; Rambaut, A. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature 2014, 508, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Paccaud, M.F.; Couard, M.; Bürki, F.; Gerber, H.; Löhrer, J. Outbreak of influenza a/equi-2 in switzerland. Nature 1966, 2011, 101–102. [Google Scholar] [CrossRef]
- Bryant, N.A.; Rash, A.S.; Russell, C.A.; Ross, J.; Cooke, A.; Bowman, S.; MacRae, S.; Lewis, N.S.; Paillot, R.; Zanoni, R.; et al. Antigenic and genetic variations in european and north american equine influenza virus strains (H3N8) isolated from 2006 to 2007. Vet. Microbiol. 2009, 138, 41–52. [Google Scholar] [CrossRef] [PubMed]
- OIE. Expert surveillance panel on equine influenza vaccine composition-conclusions and recommendations. Off. Int. Epizoot. Bull. 2010, 44–45. [Google Scholar]
- Woodward, A.; Rash, A.S.; Medcalf, E.; Bryant, N.A.; Elton, D.M. Using epidemics to map h3 equine influenza virus determinants of antigenicity. Virology 2015, 481, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhou, H.; Liu, H.; Chan, W.; Adhikary, L.; Mahmood, K.; Lee, M.S.; Kemble, G. Two residues in the hemagglutinin of a/fujian/411/02-like influenza viruses are responsible for antigenic drift from a/panama/2007/99. Virology 2005, 336, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. Bioedit: A user-friendly biological sequence alingment editor and analysis program for windows 95/98/nt. Nucleic Acids Symp. 1999, 41, 95–98. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. Jmodeltest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with beauti and the beast 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Rambaut, A. Many-core algorithms for statistical phylogenetics. Bioinformatics 2009, 25, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. Beagle: An application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the cipres science gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8.
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 14 October 2014).
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. Spread3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clade | Period | tMRCA (HPD 95%) * |
|---|---|---|
| Group I | 1963–1969 | 1962 (1961–1963) |
| Group VIII | 1985 | 1984 (1983–1984) |
| South American Clade 1 | 1993–1996 | 1992 (1992–1993) |
| South American Clade 2 | 1997–2005 | 1997 (1996–1997) |
| Florida Clade 1 | 2012 | 2011 (2011–2012) |
| Group | Aa Substitution |
|---|---|
| Group I | K50R; T131N; S149N; S265G; L431I; A476T; L496V |
| Group I and A/eq/Uruguay/1/1963 | G82; R323; I347; G381 |
| South American clade 1 | N312K |
| South American clade 2 | S92N |
| Florida clade 1 | K-14A; M70V |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olguin Perglione, C.; Golemba, M.D.; Torres, C.; Barrandeguy, M. Molecular Epidemiology and Spatio-Temporal Dynamics of the H3N8 Equine Influenza Virus in South America. Pathogens 2016, 5, 61. https://doi.org/10.3390/pathogens5040061
Olguin Perglione C, Golemba MD, Torres C, Barrandeguy M. Molecular Epidemiology and Spatio-Temporal Dynamics of the H3N8 Equine Influenza Virus in South America. Pathogens. 2016; 5(4):61. https://doi.org/10.3390/pathogens5040061
Chicago/Turabian StyleOlguin Perglione, Cecilia, Marcelo D. Golemba, Carolina Torres, and Maria Barrandeguy. 2016. "Molecular Epidemiology and Spatio-Temporal Dynamics of the H3N8 Equine Influenza Virus in South America" Pathogens 5, no. 4: 61. https://doi.org/10.3390/pathogens5040061