RTA Occupancy of the Origin of Lytic Replication during Murine Gammaherpesvirus 68 Reactivation from B Cell Latency

 , and
, and
Abstract
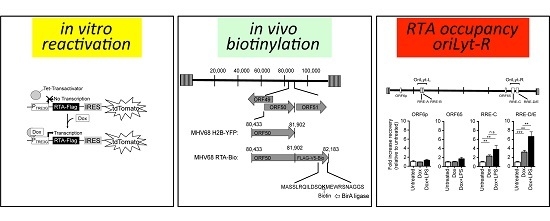
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
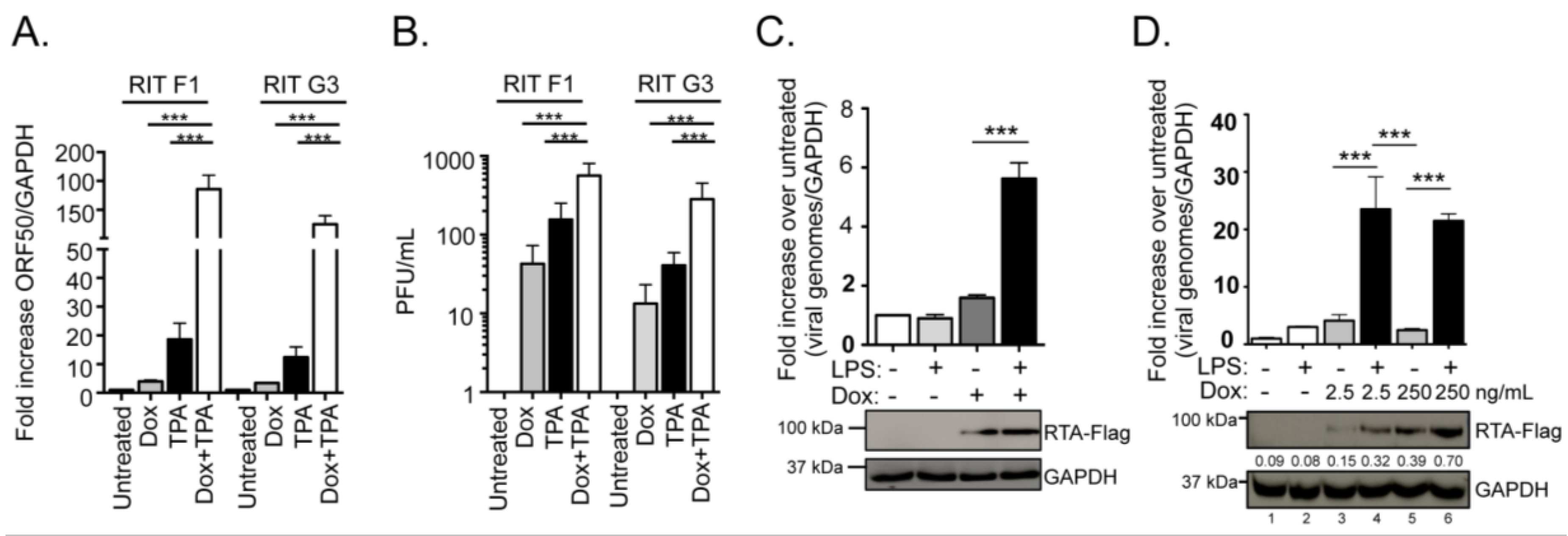
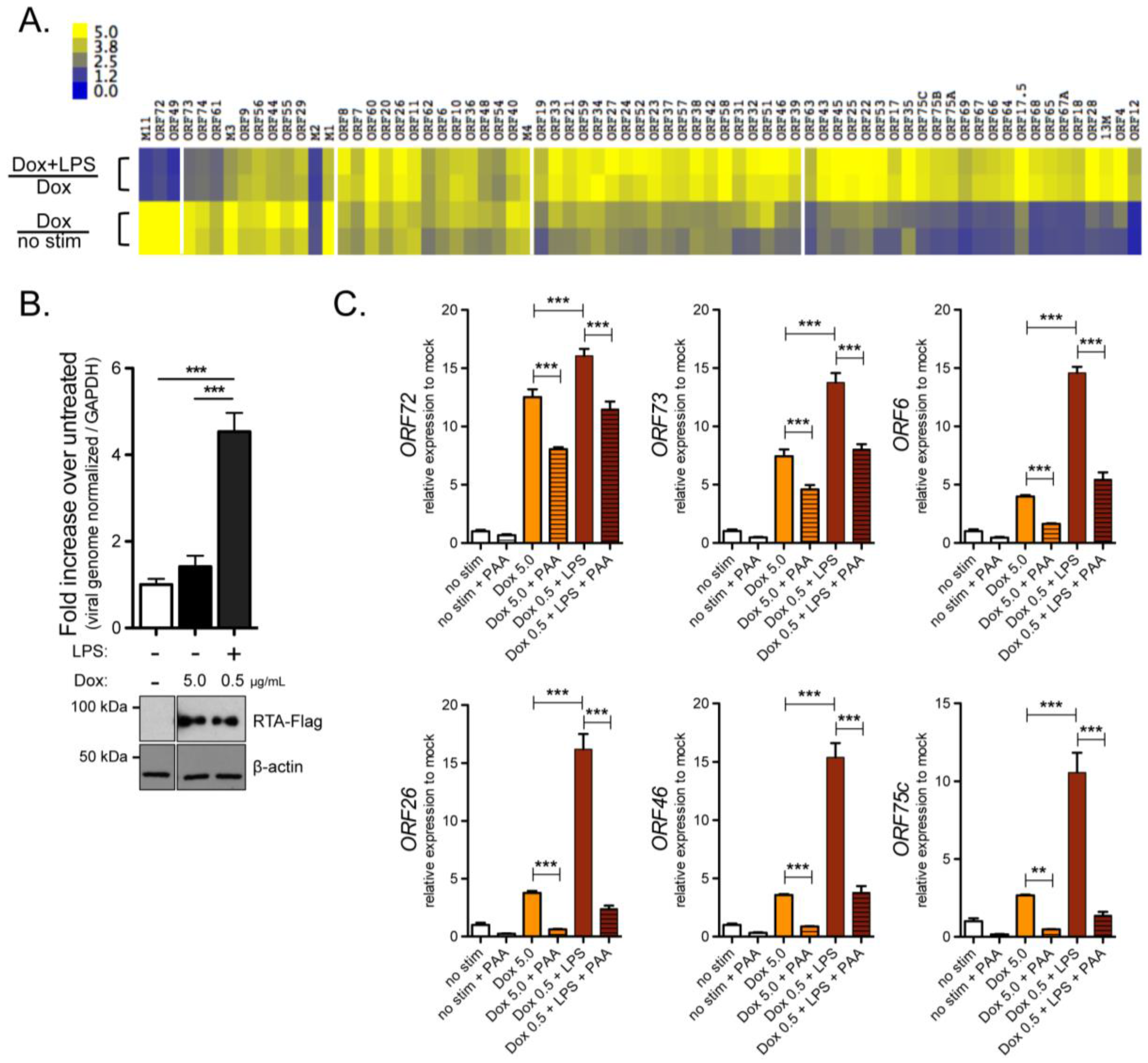
2.1. Characterization of the Response of RTA Inducible A20-HE2 Latent B Cell Lines to Lipopolysaccharide
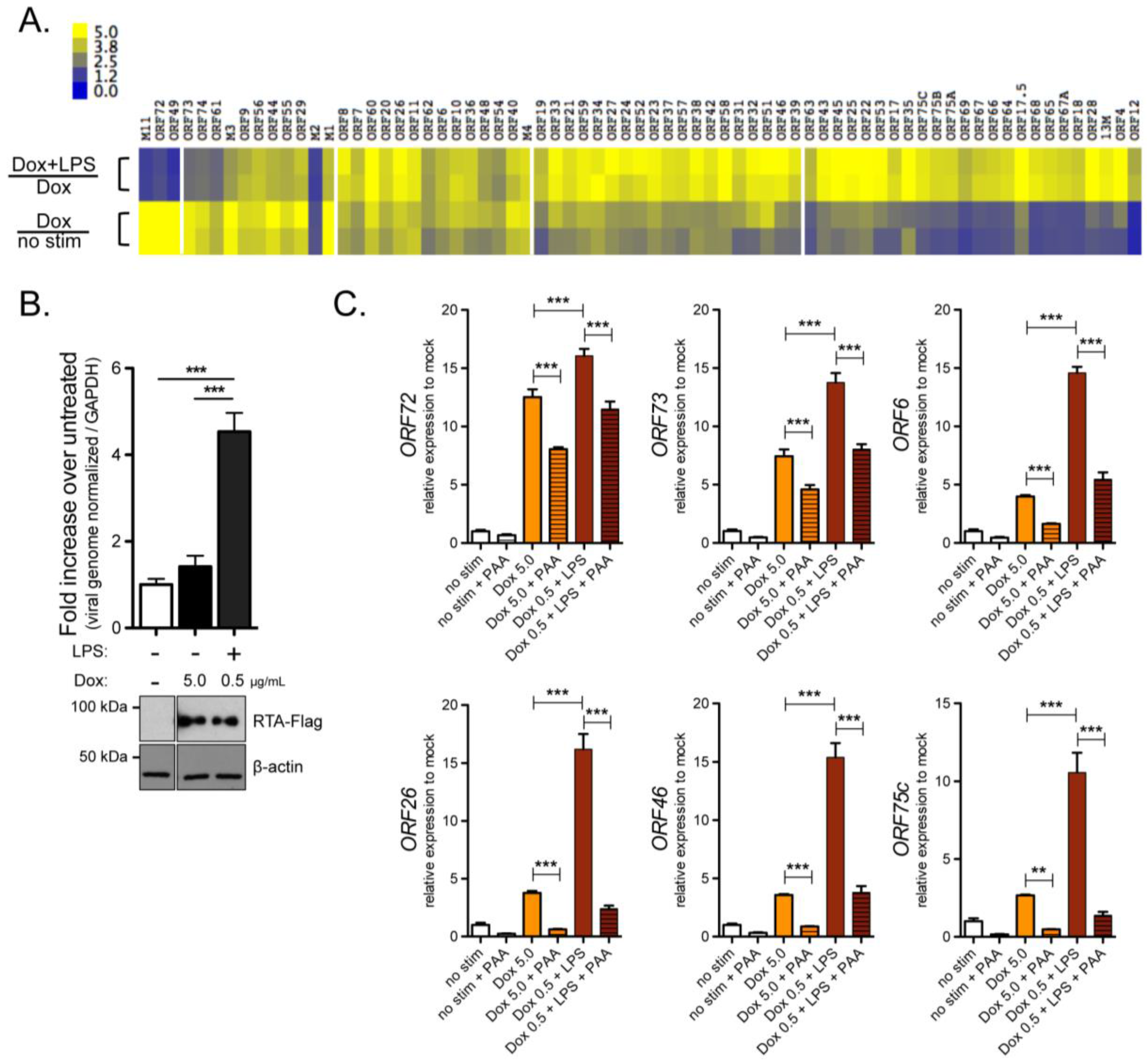
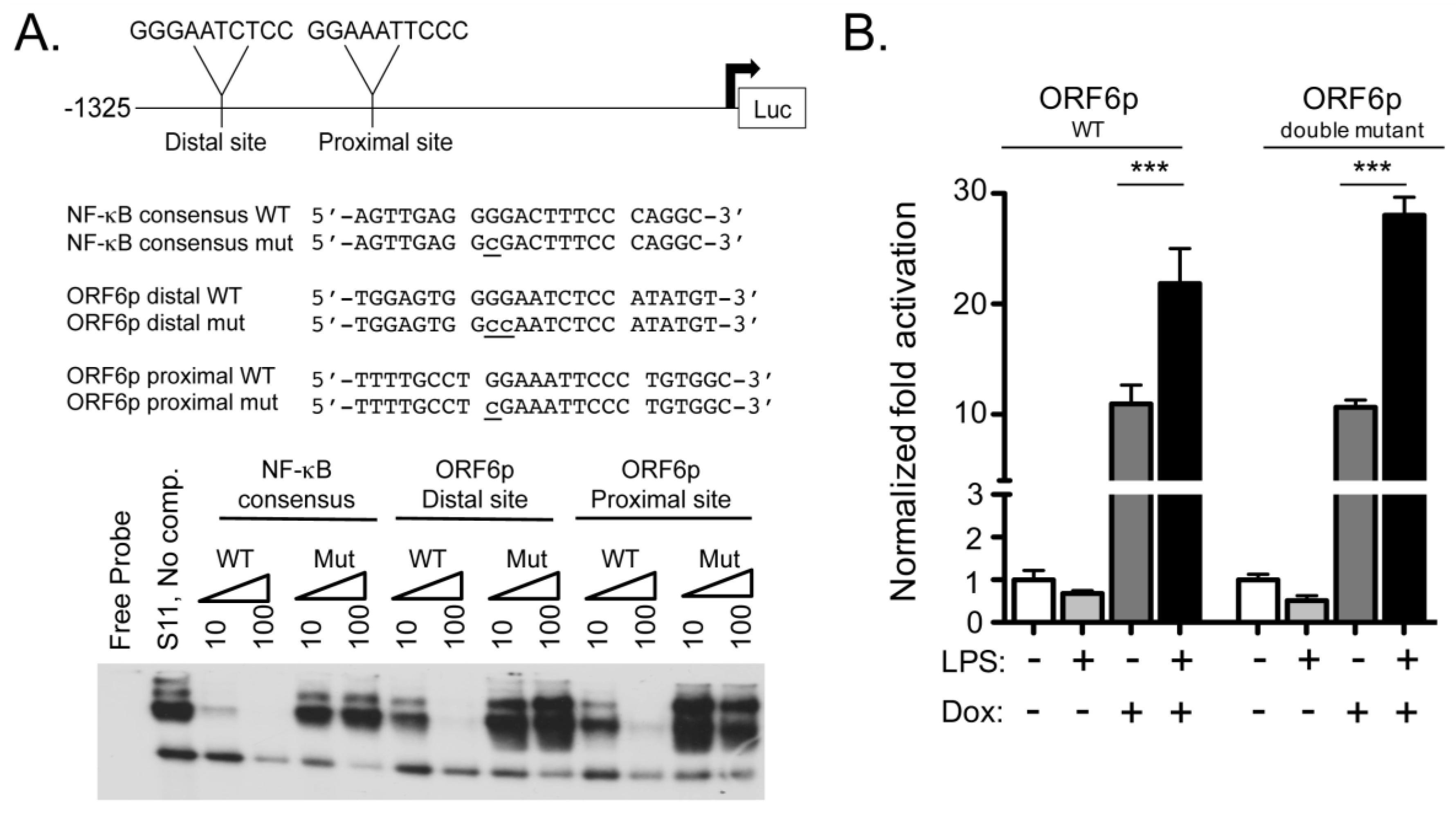
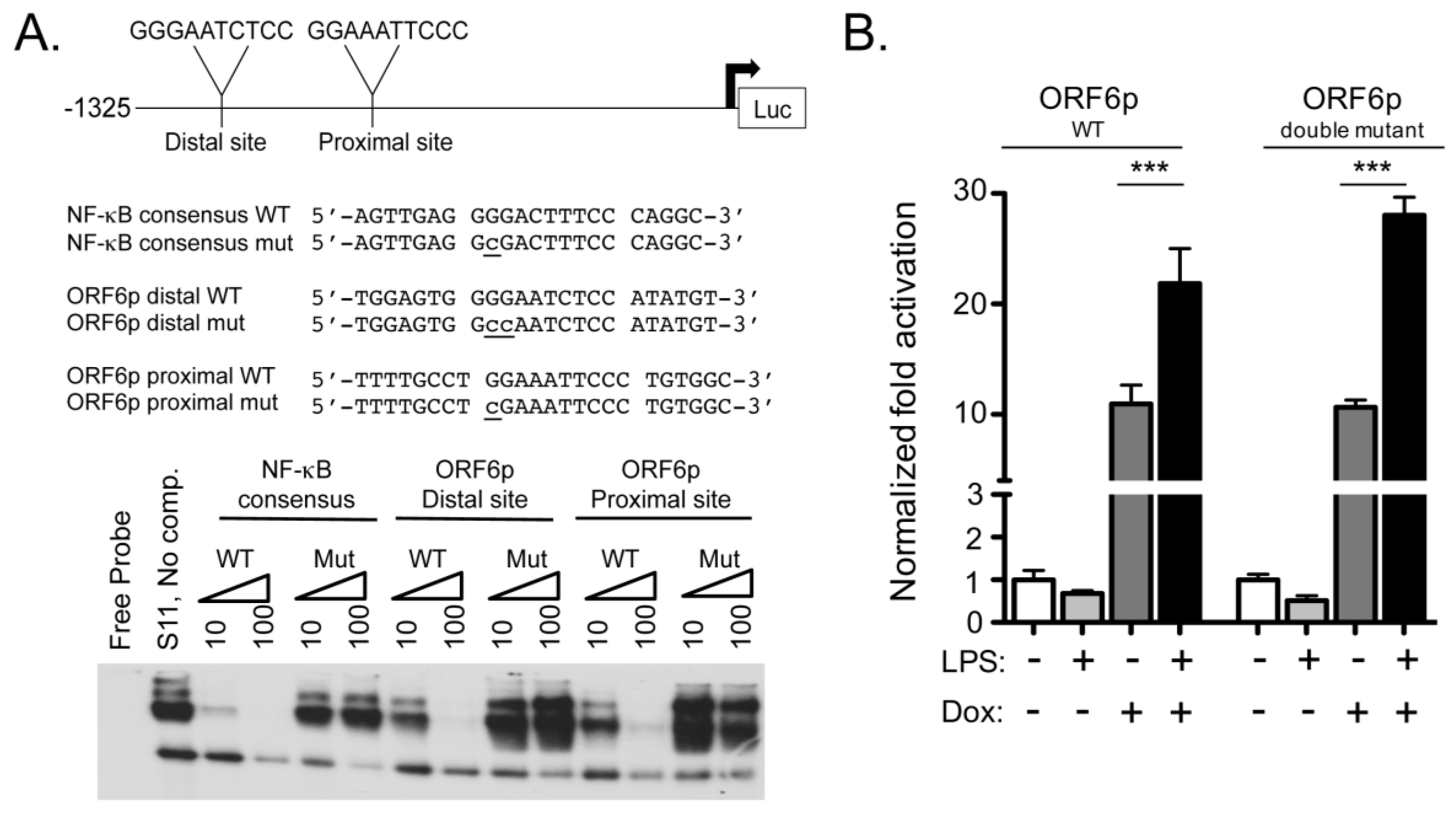
2.2. ORF6 Transactivation by RTA Is Independent of NF-κB Recognition Sites
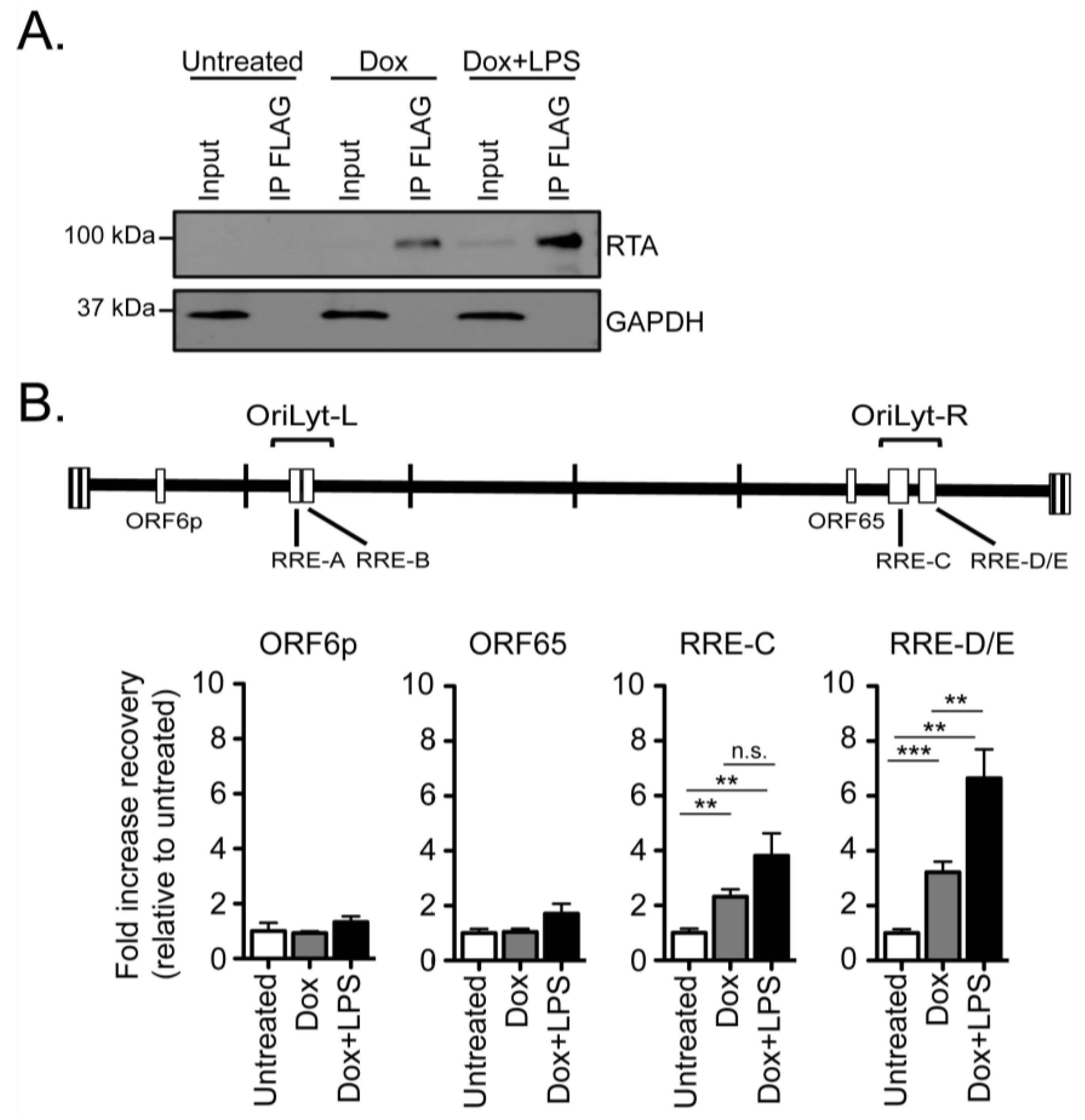
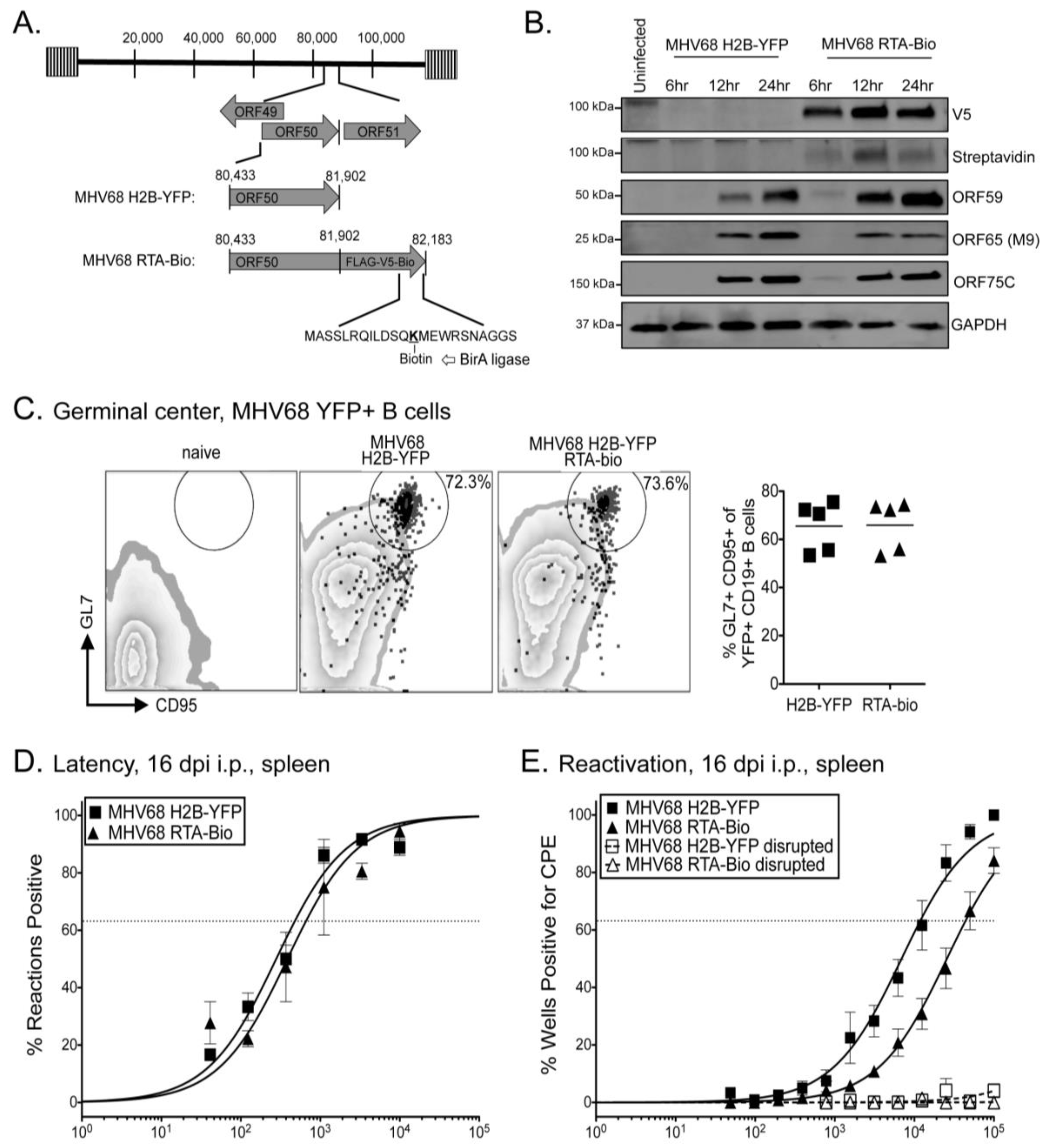
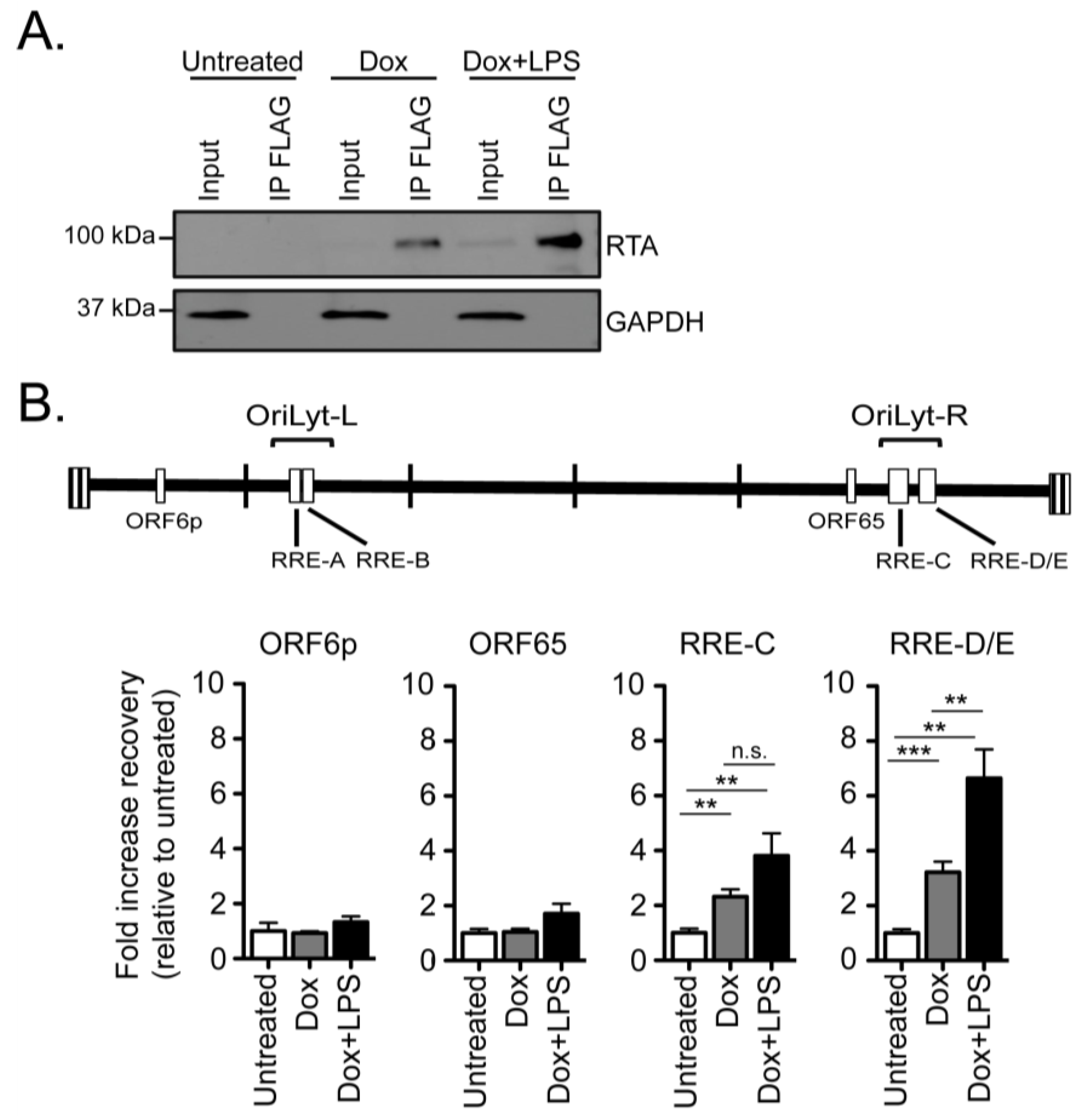
2.3. RTA Occupancy at the Right Origin of Lytic Replication upon Induced Reactivation from Latency in Cultured B Cells
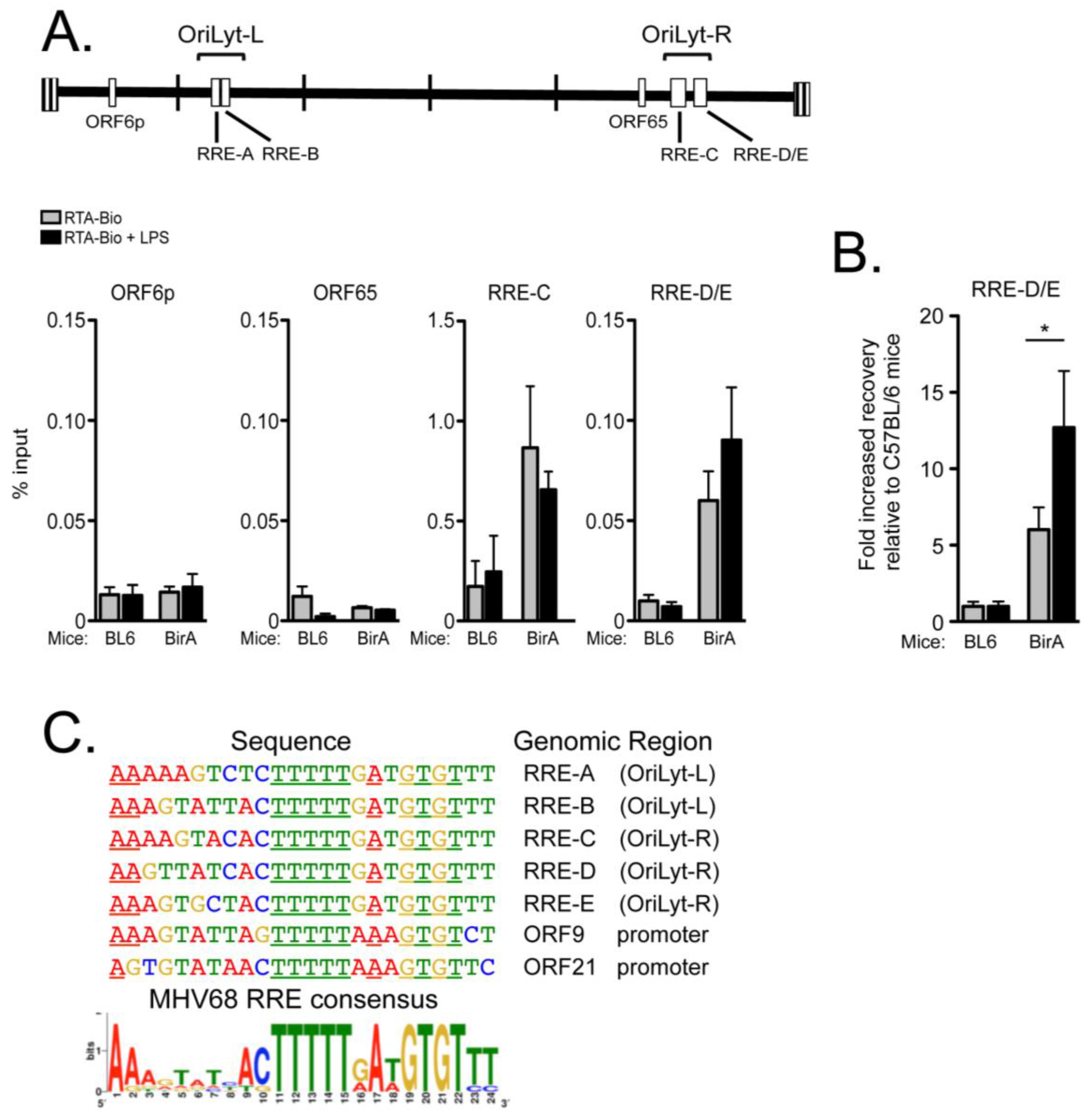
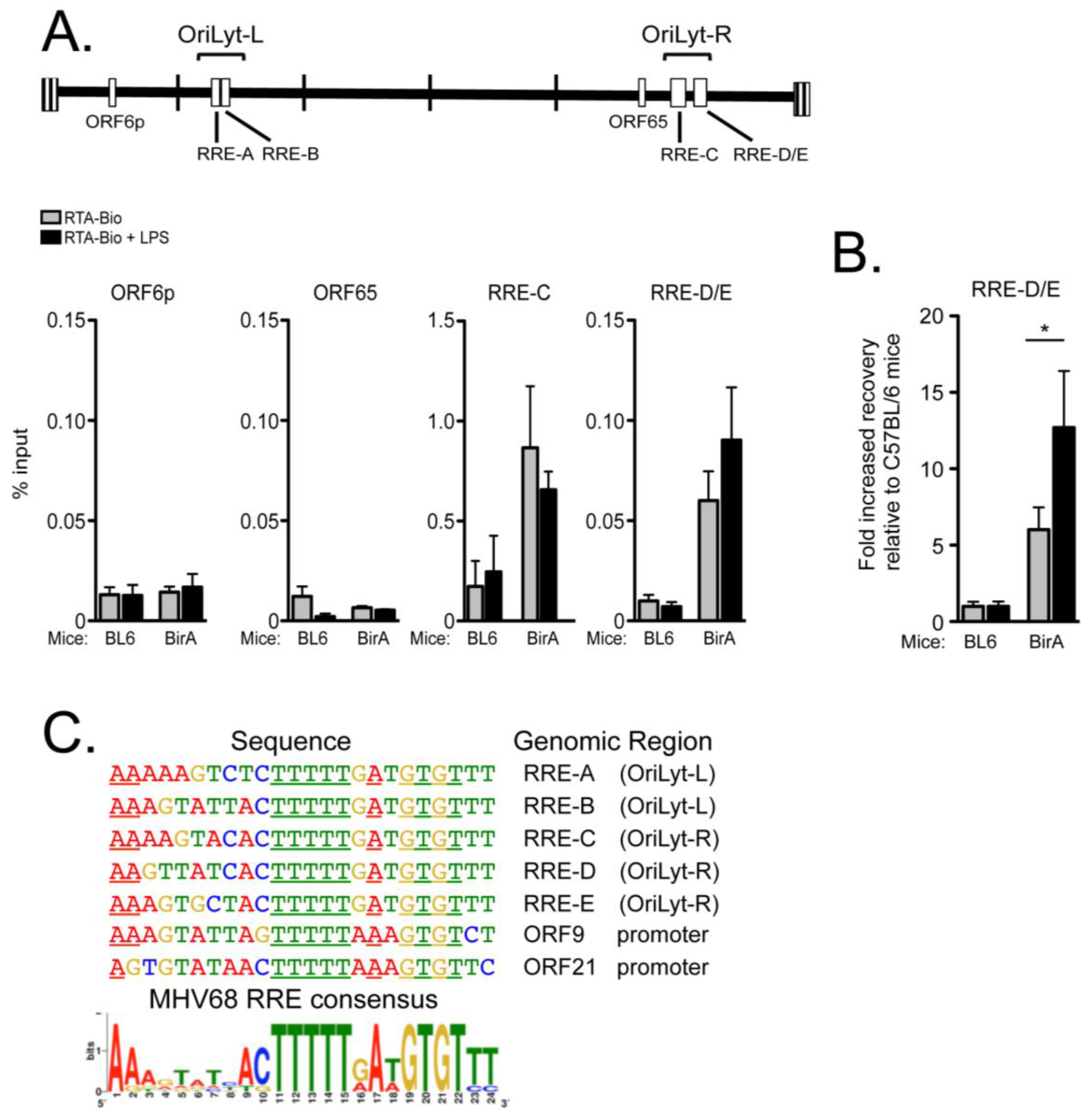
2.4. RTA Complexes with the Right Origin of Lytic Replication in Infected Primary Splenocytes upon Explant Reactivation
3. Discussion
3.1. Role of NF-κB Subunits
3.2. TLR4 Signaling Enhances Reactivation from Latency
3.3. RTA Occupancy of Origins of Lytic Replication
3.4. Conclusions
4. Materials and Methods
4.1. Cells and Mice
4.2. Plasmids
4.3. Generation of Doxycycline Inducible B Cell Lines
4.4. Generation of Recombinant Viruses
4.5. Analysis of Recombinant Viral BAC DNA
4.6. Viruses and Plaque Assays
4.7. Antibodies and Immunoblot Analysis
4.8. Quantitative PCR
4.9. RNA Seq and Data Analysis
4.10. Quantitative RT-PCR
4.11. Electrophoretic Mobility Shift Assays
4.12. Luciferase Reporter Assays
4.13. Chromatin Immunoprecipitation
4.14. Infections and Organ Harvests
4.15. Limiting Dilution PCR Detection of MHV68 Genome Positive Cells
4.16. Limiting Dilution Ex Vivo Reactivation Assay
4.17. Motif Analysis
4.18. Statistical Analyses
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heslop, H.E. Biology and treatment of epstein-barr virus-associated non-hodgkin lymphomas. ASH Educ. Progr. Book. 2005, 260, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Cesarman, E.; Spina, M.; Gloghini, A.; Schulz, T.F. Hiv-associated lymphomas and gamma-herpesviruses. Blood 2009, 113, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Cabras, A.; Elia, G. The germinal centre-derived lymphomas seen through their cellular microenvironment. Br. J. Haematol. 2009, 145, 468–480. [Google Scholar] [CrossRef] [PubMed]
- DePond, W.; Said, J.W.; Tasaka, T.; de Vos, S.; Kahn, D.; Cesarman, E.; Knowles, D.M.; Koeffler, H.P. Kaposi’s sarcoma-associated herpesvirus and human herpesvirus 8 (KSHV/hhv8)-associated lymphoma of the bowel. Report of two cases in hiv-positive men with secondary effusion lymphomas. Am. J. Surg. Pathol. 1997, 21, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Nash, A.A.; Dutia, B.M.; Stewart, J.P.; Davison, A.J. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2001, 356, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, E.; Mandal, P.; Speck, S.H. Pathogenesis and host control of gammaherpesviruses: Lessons from the mouse. Annu. Rev. Immunol. 2011, 29, 351–397. [Google Scholar] [CrossRef] [PubMed]
- Simas, J.P.; Efstathiou, S. Murine gammaherpesvirus 68: A model for the study of gammaherpesvirus pathogenesis. Trends Microbiol. 1998, 6, 276–282. [Google Scholar] [CrossRef]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the gamma-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Blackman, M.A.; Flano, E.; Usherwood, E.; Woodland, D.L. Murine gamma-herpesvirus-68: A mouse model for infectious mononucleosis? Mol. Med. Today 2000, 6, 488–490. [Google Scholar] [CrossRef]
- Wu, T.T.; Usherwood, E.J.; Stewart, J.P.; Nash, A.A.; Sun, R. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 2000, 74, 3659–3667. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. KSHV RTA promoter specification and viral reactivation. Front. Microbiol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. KSHV reactivation and novel implications of protein isomerization on lytic switch control. Viruses 2015, 7, 72–109. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, I.V.; Virgin, H.W.; Speck, S.H. Disruption of gammaherpesvirus 68 gene 50 demonstrates that RTA is essential for virus replication. J. Virol. 2003, 77, 5731–5739. [Google Scholar] [CrossRef] [PubMed]
- Ragoczy, T.; Heston, L.; Miller, G. The epstein-barr virus RTA protein activates lytic cycle genes and can disrupt latency in b lymphocytes. J. Virol. 1998, 72, 7978–7984. [Google Scholar] [PubMed]
- Allen, R.D., 3rd; DeZalia, M.N.; Speck, S.H. Identification of an RTA responsive promoter involved in driving gammahv68 v-cyclin expression during virus replication. Virology 2007, 365, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Qi, J.; Gong, D.; Han, C.; Deng, H. Replication and transcription activator (RTA) of murine gammaherpesvirus 68 binds to an RTA-responsive element and activates the expression of orf18. J. Virol. 2011, 85, 11338–11350. [Google Scholar] [CrossRef] [PubMed]
- Noh, C.W.; Cho, H.J.; Kang, H.R.; Jin, H.Y.; Lee, S.; Deng, H.; Wu, T.T.; Arumugaswami, V.; Sun, R.; Song, M.J. The virion-associated open reading frame 49 of murine gammaherpesvirus 68 promotes viral replication both in vitro and in vivo as a derepressor of RTA. J. Virol. 2012, 86, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, B.M.; Soni, T.; Wakeman, B.S.; Speck, S.H. The murine gammaherpesvirus immediate-early RTA synergizes with irf4, targeting expression of the viral m1 superantigen to plasma cells. PLoS Pathog. 2014, 10, e1004302. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, I.; Lin, C.Y.; Speck, S.H. Murine gammaherpesvirus 68 RTA-dependent activation of the gene 57 promoter. Virology 2005, 333, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Han, C.; Gong, D.; Liu, P.; Zhou, S.; Deng, H. Murine gammaherpesvirus 68 orf48 is an RTA-responsive gene product and functions in both viral lytic replication and latency during in vivo infection. J. Virol. 2015, 89, 5788–5800. [Google Scholar] [CrossRef] [PubMed]
- Damania, B.; Jeong, J.H.; Bowser, B.S.; DeWire, S.M.; Staudt, M.R.; Dittmer, D.P. Comparison of the RTA/orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 2004, 78, 5491–5499. [Google Scholar] [CrossRef] [PubMed]
- Rickabaugh, T.M.; Brown, H.J.; Wu, T.T.; Song, M.J.; Hwang, S.; Deng, H.; Mitsouras, K.; Sun, R. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 RTA reactivates murine gammaherpesvirus 68 from latency. J. Virol. 2005, 79, 3217–3222. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappab family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The ikk complex, a central regulator of NF-kappab activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.J.; Song, M.J.; Deng, H.; Wu, T.T.; Cheng, G.; Sun, R. Nf-κB inhibits gammaherpesvirus lytic replication. J. Virol. 2003, 77, 8532–8540. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.T.; Moser, J.M.; Dickerson, S.M.; Speck, S.H. Inhibition of NF-kappab activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 2007, 3, e11. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Flano, E.; Woodland, D.L.; Lund, F.E.; Randall, T.D.; Blackman, M.A. Maintenance of long term gamma-herpesvirus b cell latency is dependent on cd40-mediated development of memory b cells. J. Immunol. 2003, 171, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Willer, D.O.; Speck, S.H. Establishment and maintenance of long-term murine gammaherpesvirus 68 latency in b cells in the absence of cd40. J. Virol. 2005, 79, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Frederico, B.; May, J.S.; Efstathiou, S.; Stevenson, P.G. Baff receptor deficiency limits gammaherpesvirus infection. J. Virol. 2014, 88, 3965–3975. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.T.; Collins, C.M.; Gargano, L.M.; Speck, S.H. Nf-kappab p50 plays distinct roles in the establishment and control of murine gammaherpesvirus 68 latency. J. Virol. 2009, 83, 4732–4748. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Feng, H.; Sun, Q.; Li, H.; Wu, T.T.; Sun, R.; Tibbetts, S.A.; Chen, Z.J.; Feng, P. Murine gamma-herpesvirus 68 hijacks mavs and ikkbeta to initiate lytic replication. PLoS Pathog. 2010, 6, e1001001. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; He, Z.; Durakoglugil, D.; Arneson, L.; Shen, Y.; Feng, P. Murine gammaherpesvirus 68 evades host cytokine production via replication transactivator-induced rela degradation. J. Virol. 2012, 86, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Cieniewicz, B.; Santana, A.L.; Minkah, N.; Krug, L.T. Interplay of murine gammaherpesvirus 68 with NF-kappab signaling of the host. Front. Microbiol. 2016, 7, 1202. [Google Scholar] [CrossRef] [PubMed]
- Gargano, L.M.; Forrest, J.C.; Speck, S.H. Signaling through toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J. Virol. 2009, 83, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Haas, F.; Yamauchi, K.; Murat, M.; Bernasconi, M.; Yamanaka, N.; Speck, R.F.; Nadal, D. Activation of NF-kappab via endosomal toll-like receptor 7 (tlr7) or tlr9 suppresses murine herpesvirus 68 reactivation. J. Virol. 2014, 88, 10002–10012. [Google Scholar] [CrossRef] [PubMed]
- Gargano, L.M.; Moser, J.M.; Speck, S.H. Role for myd88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 2008, 82, 3853–3863. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, S.A.; Suarez, F.; Steed, A.L.; Simmons, J.A.; Virgin, H.W.t. A gamma-herpesvirus deficient in replication establishes chronic infection in vivo and is impervious to restriction by adaptive immune cells. Virology 2006, 353, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.M.; Upton, J.W.; Gray, K.S.; Speck, S.H. Ex vivo stimulation of b cells latently infected with gammaherpesvirus 68 triggers reactivation from latency. J. Virol. 2005, 79, 5227–5231. [Google Scholar] [CrossRef] [PubMed]
- Forrest, J.C.; Speck, S.H. Establishment of b-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 2008, 82, 7688–7699. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.Y.; Zhi, J.; Santana, A.; Khan, S.; Salinas, E.; Forrest, J.C.; Zheng, Y.; Jaggi, S.; Leatherwood, J.; Krug, L.T. Tiled microarray identification of novel viral transcript structures and distinct transcriptional profiles during two modes of productive murine gammaherpesvirus 68 infection. J. Virol. 2012, 86, 4340–4357. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Walsh, D.; Harbell, J.; Wilson, A.C.; Mohr, I. Activation of host translational control pathways by a viral developmental switch. PLoS Pathog. 2009, 5, e1000334. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.W.; Powell, K.L.; Kellam, P.; Alber, D.G. Gammaherpesvirus lytic gene expression as characterized by DNA array. J. Virol. 2002, 76, 6244–6256. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, B.; Dutia, B.M.; Roberts, K.L.; Garcia-Ramirez, J.J.; Dickinson, P.; Stewart, J.P.; Ghazal, P.; Roy, D.J.; Nash, A.A. Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J. Gen. Virol. 2003, 84, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Guzman, D.; Rickabaugh, T.; Wu, T.-T.; Brown, H.; Cole, S.; Song, M.J.; Tong, L.; Sun, R. Transcription program of murine gammaherpesvirus 68. J. Virol. 2003, 77, 10488–10503. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Willert, E.K.; Virgin, H.W. Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe 2010, 7, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Izumiya, C.; Hsia, D.; Ellison, T.J.; Luciw, P.A.; Kung, H.J. Nf-kappab serves as a cellular sensor of kaposi’s sarcoma-associated herpesvirus latency and negatively regulates k-RTA by antagonizing the rbp-jkappa coactivator. J. Virol. 2009, 83, 4435–4446. [Google Scholar] [CrossRef] [PubMed]
- Howard, P.K.; Shaw, J.; Otsuka, A.J. Nucleotide sequence of the bira gene encoding the biotin operon repressor and biotin holoenzyme synthetase functions of escherichia coli. Gene 1985, 35, 321–331. [Google Scholar] [CrossRef]
- de Boer, E.; Rodriguez, P.; Bonte, E.; Krijgsveld, J.; Katsantoni, E.; Heck, A.; Grosveld, F.; Strouboulis, J. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 7480–7485. [Google Scholar] [CrossRef] [PubMed]
- Driegen, S.; Ferreira, R.; van Zon, A.; Strouboulis, J.; Jaegle, M.; Grosveld, F.; Philipsen, S.; Meijer, D. A generic tool for biotinylation of tagged proteins in transgenic mice. Transgenic Res. 2005, 14, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Qi, J.; Gong, D.-Y.; Deng, J.-W.; Deng, H.-Y. Identification of an RTA responsive element in murine gammaherpesvirus-68 genome. Prog. Biochem. Biophys. 2011, 38, 936–944. [Google Scholar] [CrossRef]
- Grossmann, C.; Ganem, D. Effects of nfkappab activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 2008, 375, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Sgarbanti, M.; Arguello, M.; tenOever, B.R.; Battistini, A.; Lin, R.; Hiscott, J. A requirement for NF-kappab induction in the production of replication-competent hhv-8 virions. Oncogene 2004, 23, 5770–5780. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef] [PubMed]
- Kishore, N.; Sommers, C.; Mathialagan, S.; Guzova, J.; Yao, M.; Hauser, S.; Huynh, K.; Bonar, S.; Mielke, C.; Albee, L.; et al. A selective ikk-2 inhibitor blocks NF-kappa b-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J. Biol. Chem. 2003, 278, 32861–32871. [Google Scholar] [CrossRef] [PubMed]
- Mounce, B.C.; Tsan, F.C.; Kohler, S.; Cirillo, L.A.; Tarakanova, V.L. Dynamic association of gammaherpesvirus DNA with core histone during de novo lytic infection of primary cells. Virology 2011, 421, 167–172. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Xu, Y.; Cei, S.A.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J. Virol. 2002, 76, 7890–7896. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Li, H.; Wang, Y.; Zhu, F.X.; Kudchodkar, S.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus lytic origin (ori-lyt)-dependent DNA replication: Identification of the ori-lyt and association of k8 bzip protein with the origin. J. Virol. 2003, 77, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, W.; Sugden, B. Identification and characterization of orilyt, a lytic origin of DNA replication of epstein-barr virus. Cell 1988, 55, 427–433. [Google Scholar] [CrossRef]
- Deng, H.; Chu, J.T.; Park, N.H.; Sun, R. Identification of cis sequences required for lytic DNA replication and packaging of murine gammaherpesvirus 68. J. Virol. 2004, 78, 9123–9131. [Google Scholar] [CrossRef] [PubMed]
- Sattler, C.; Steer, B.; Adler, H. Multiple lytic origins of replication are required for optimal gammaherpesvirus fitness in vitro and in vivo. PLoS Pathog. 2016, 12, e1005510. [Google Scholar] [CrossRef] [PubMed]
- Fixman, E.D.; Hayward, G.S.; Hayward, S.D. Replication of epstein-barr virus orilyt: Lack of a dedicated virally encoded origin-binding protein and dependence on zta in cotransfection assays. J. Virol. 1995, 69, 2998–3006. [Google Scholar] [PubMed]
- Wu, F.Y.; Ahn, J.H.; Alcendor, D.J.; Jang, W.J.; Xiao, J.; Hayward, S.D.; Hayward, G.S. Origin-independent assembly of kaposi’s sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the orf-k8 protein and cellular pml. J. Virol. 2001, 75, 1487–1506. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Chen, H.; Shamay, M.; Zheng, Q.; Hayward, G.S. Early activation of the kaposi’s sarcoma-associated herpesvirus RTA, rap, and mta promoters by the tetradecanoyl phorbol acetate-induced ap1 pathway. J. Virol. 2004, 78, 4248–4267. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Cei, S.A.; Papouskova, I.; Tarrant, M.; Pari, G.S. Amplification of the kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting at-rich region and an orf50 response element and the trans-acting factors orf50 (k-RTA) and k8 (k-bzip). Virology 2004, 318, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Susilarini, N.K.; Pari, G.S. Interaction of kaposi’s sarcoma-associated herpesvirus orf59 with orilyt is dependent on binding with k-RTA. J. Virol. 2011, 85, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Ellison, T.J.; Izumiya, Y.; Izumiya, C.; Luciw, P.A.; Kung, H.J. A comprehensive analysis of recruitment and transactivation potential of k-RTA and k-bzip during reactivation of kaposi’s sarcoma-associated herpesvirus. Virology 2009, 387, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Sharma, N.; Murakami, M.; Robertson, E.S. Induction of kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: A novel mechanism for establishment of latency. J. Virol. 2005, 79, 7453–7465. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; Dittmer, D.P. Quantitative analysis of the bidirectional viral g-protein-coupled receptor and lytic latency-associated nuclear antigen promoter of kaposi’s sarcoma-associated herpesvirus. J. Virol. 2012, 86, 9683–9695. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.D., 3rd; Dickerson, S.; Speck, S.H. Identification of spliced gammaherpesvirus 68 lana and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J. Virol. 2006, 80, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Usherwood, E.J.; Stewart, J.P.; Nash, A.A. Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J. Virol. 1996, 70, 6516–6518. [Google Scholar] [PubMed]
- Lukac, D.M.; Garibyan, L.; Kirshner, J.R.; Palmeri, D.; Ganem, D. DNA binding by kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 2001, 75, 6786–6799. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Williams, K.J.; Saccani, S.; Zhou, L.; Hoffmann, A.; Ghosh, G.; Gerondakis, S.; Natoli, G.; Smale, S.T. A c-rel subdomain responsible for enhanced DNA-binding affinity and selective gene activation. Genes Dev. 2005, 19, 2138–2151. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Meissner, A.; Dillon, C.P.; McManus, M.; Sharp, P.A.; Van Parijs, L.; Jaenisch, R.; Jacks, T. Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl. Acad. Sci. USA 2004, 101, 10380–10385. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.M.; Speck, S.H. Tracking murine gammaherpesvirus 68 infection of germinal center b cells in vivo. PLoS ONE 2012, 7, e33230. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, U.; Schlosser, A.; Muller, K.M.; Schafer, A.; Katava, N.; Baumeister, R.; Schulze, E. Snavi—A new tandem tag for high-affinity protein-complex purification. Nucleic Acids Res. 2010, 38, e91. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Weck, K.E.; Kim, S.S.; Virgin, H.I.; Speck, S.H. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 1999, 73, 4651–4661. [Google Scholar] [PubMed]
- Upton, J.W.; van Dyk, L.F.; Speck, S.H. Characterization of murine gammaherpesvirus 68 v-cyclin interactions with cellular cdks. Virology 2005, 341, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Sun, R. Inhibition of gammaherpesvirus replication by RNA interference. J. Virol. 2003, 77, 3301–3306. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from rna-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides accurate, fast, and bias-aware transcript expression estimates using dual-phase inference. bioRxiv 2016. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for rna-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Weck, K.E.; Barkon, M.L.; Yoo, L.I.; Speck, S.H.; Virgin, H.I. Mature b cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 1996, 70, 6775–6780. [Google Scholar] [PubMed]
- Frith, M.C.; Saunders, N.F.; Kobe, B.; Bailey, T.L. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput. Biol. 2008, 4, e1000071. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. Fimo: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. Ncbi reference sequence (refseq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana, A.L.; Oldenburg, D.G.; Kirillov, V.; Malik, L.; Dong, Q.; Sinayev, R.; Marcu, K.B.; White, D.W.; Krug, L.T. RTA Occupancy of the Origin of Lytic Replication during Murine Gammaherpesvirus 68 Reactivation from B Cell Latency. Pathogens 2017, 6, 9. https://doi.org/10.3390/pathogens6010009
Santana AL, Oldenburg DG, Kirillov V, Malik L, Dong Q, Sinayev R, Marcu KB, White DW, Krug LT. RTA Occupancy of the Origin of Lytic Replication during Murine Gammaherpesvirus 68 Reactivation from B Cell Latency. Pathogens. 2017; 6(1):9. https://doi.org/10.3390/pathogens6010009
Chicago/Turabian StyleSantana, Alexis L., Darby G. Oldenburg, Varvara Kirillov, Laraib Malik, Qiwen Dong, Roman Sinayev, Kenneth B. Marcu, Douglas W. White, and Laurie T. Krug. 2017. "RTA Occupancy of the Origin of Lytic Replication during Murine Gammaherpesvirus 68 Reactivation from B Cell Latency" Pathogens 6, no. 1: 9. https://doi.org/10.3390/pathogens6010009