
Guts Bacterial Communities of Porcellio dilatatus: Symbionts Predominance, Functional Significance and Putative Biotechnological Potential

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Gut Isolation and DNA Extraction
2.3. rRNA Gene Sequencing and Data Analysis
2.4. Functional Analysis
2.5. Statistical Analysis
3. Results
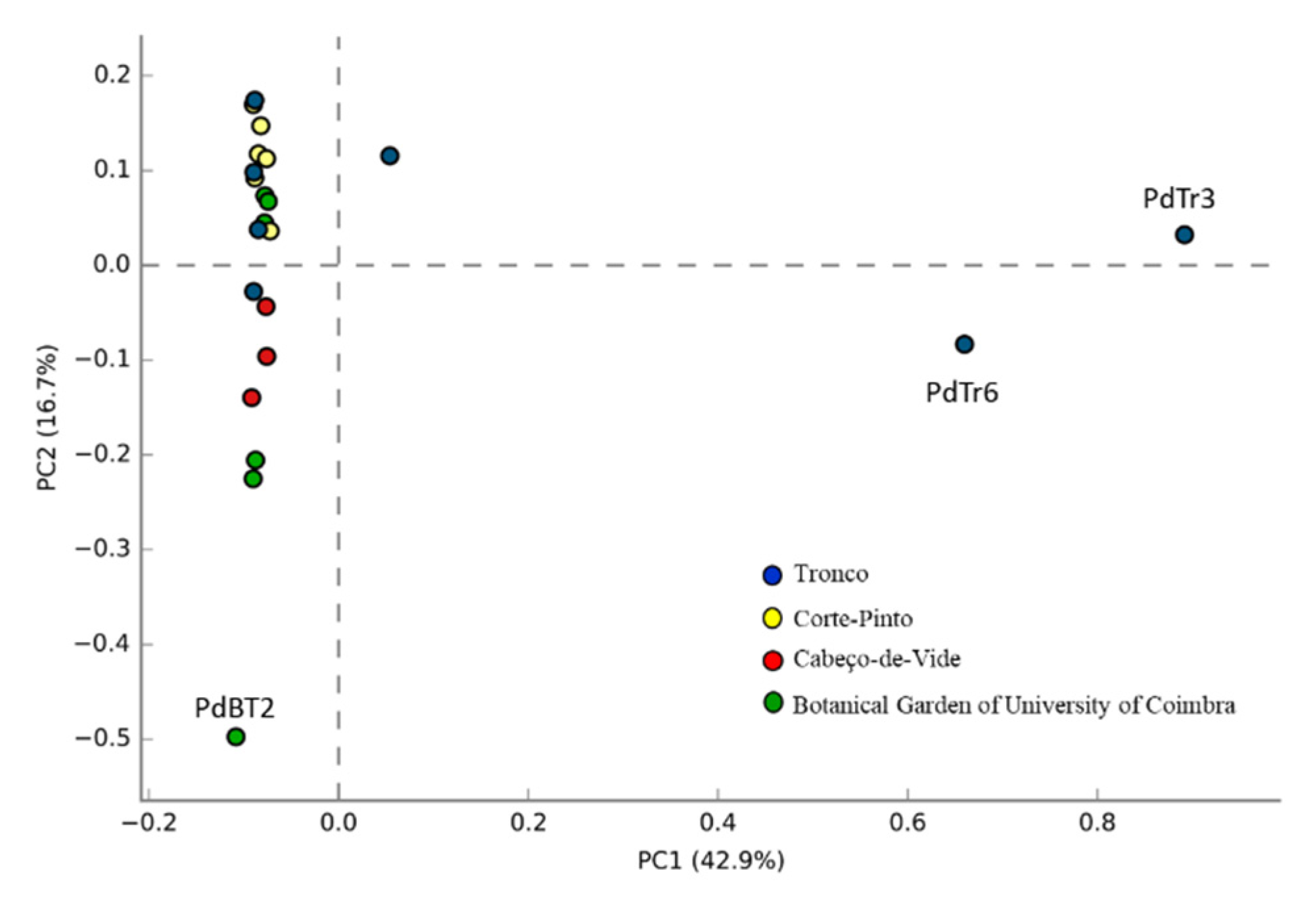
3.1. Sequencing Data Analysis
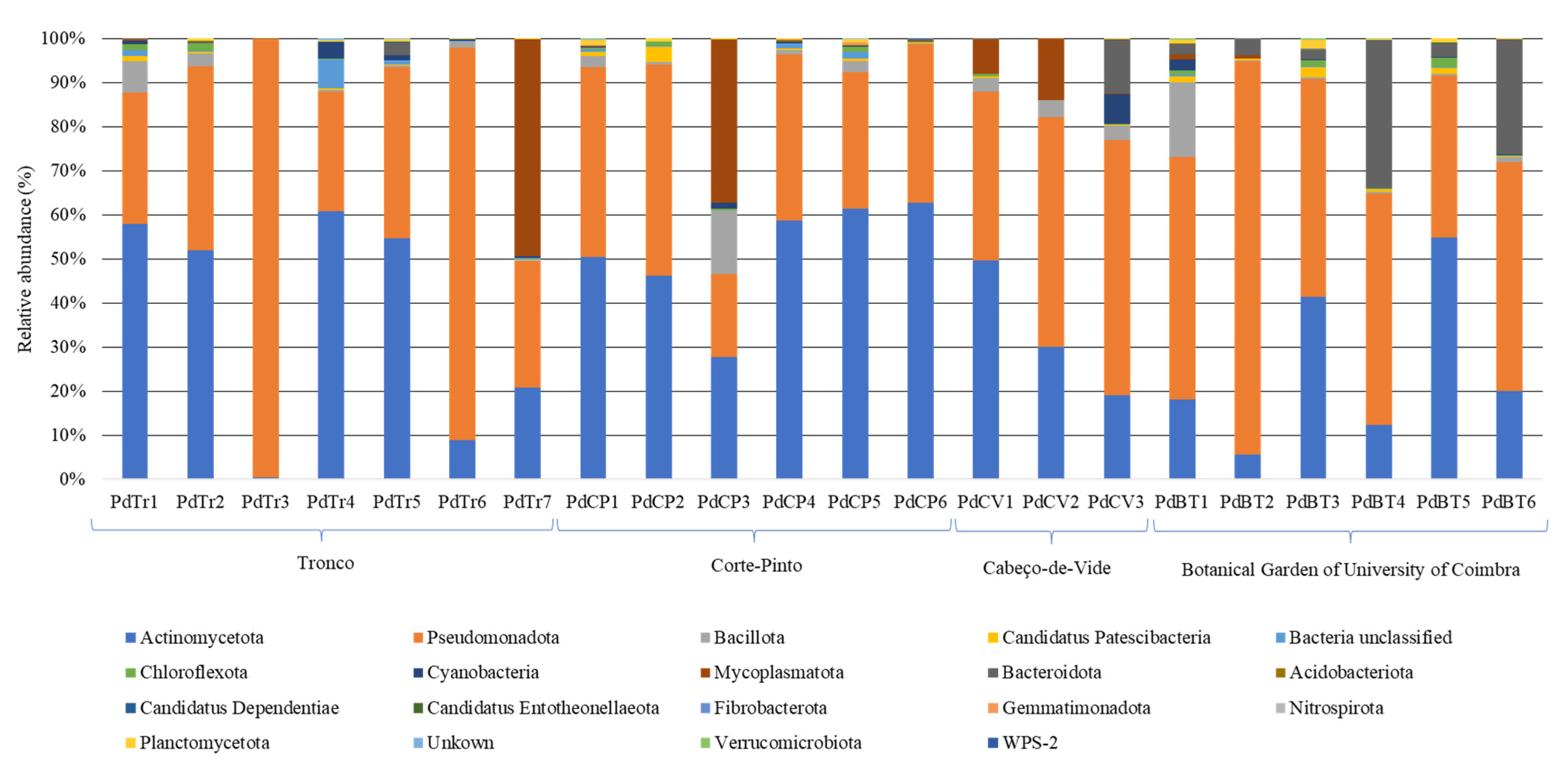
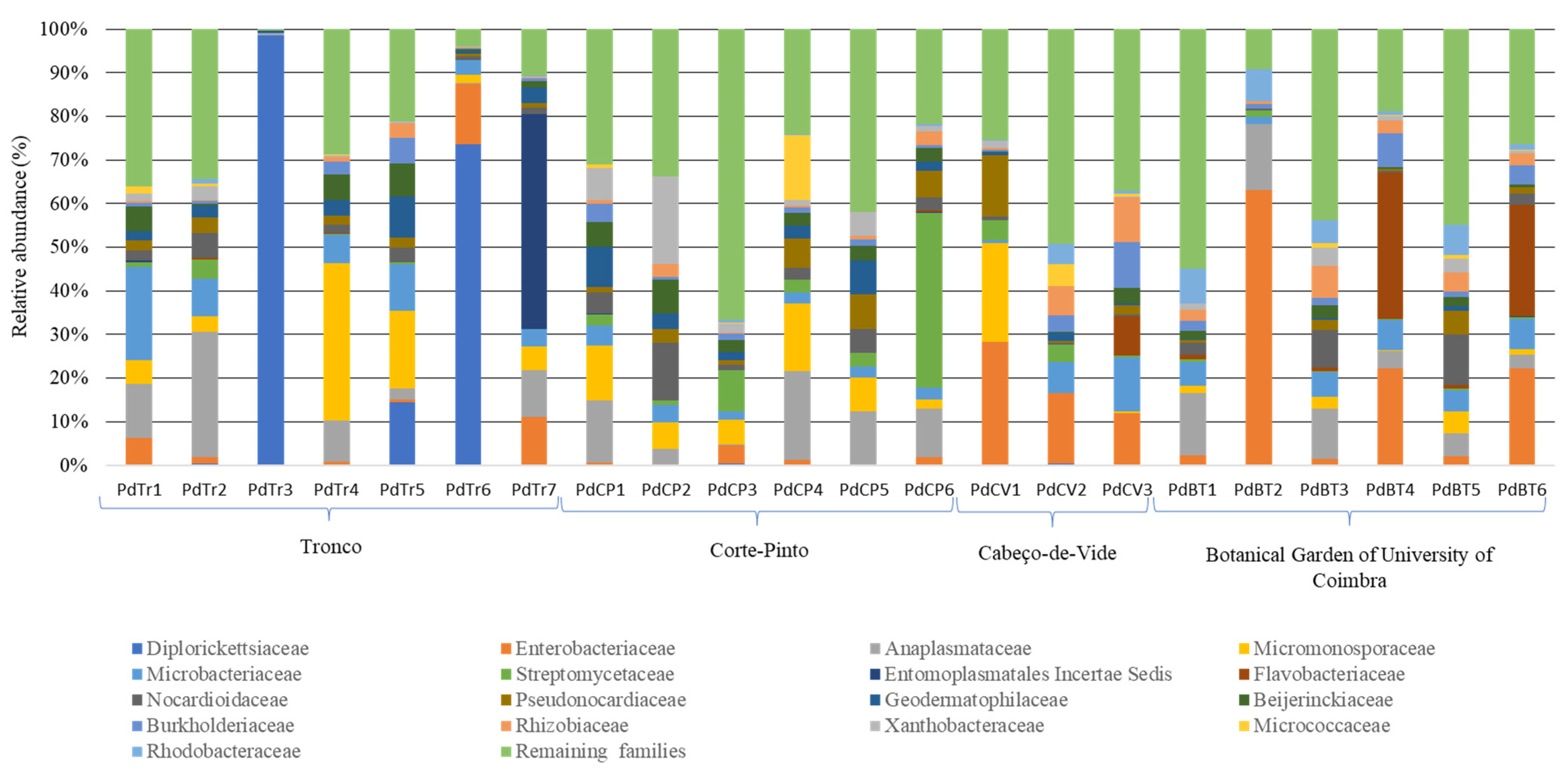
3.2. Bacterial Communities Associated to Porcellio dilatatus Guts
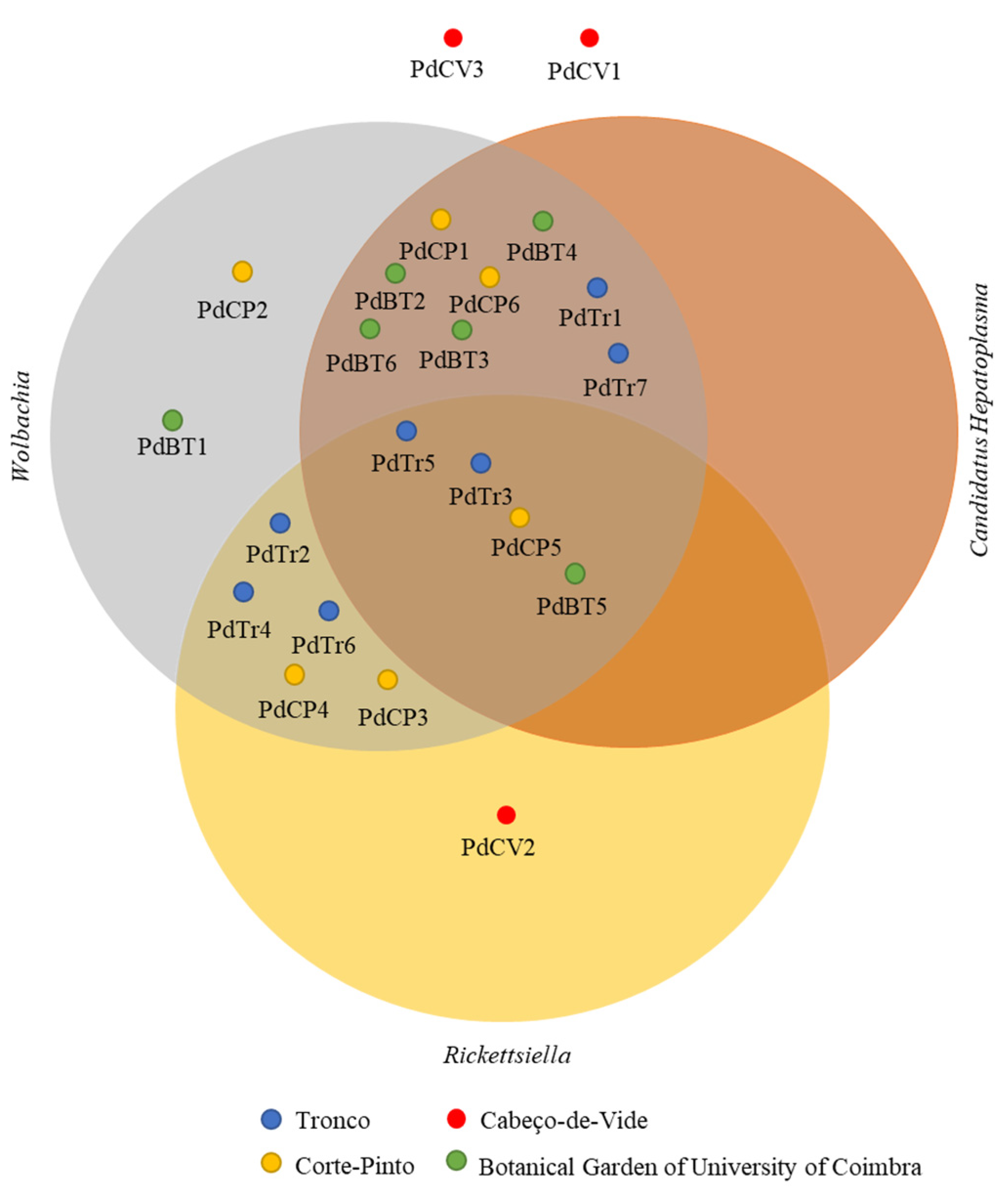
3.3. Presence of Endosymbionts and Parasites in Porcellio dilatatus Bacterial Communities
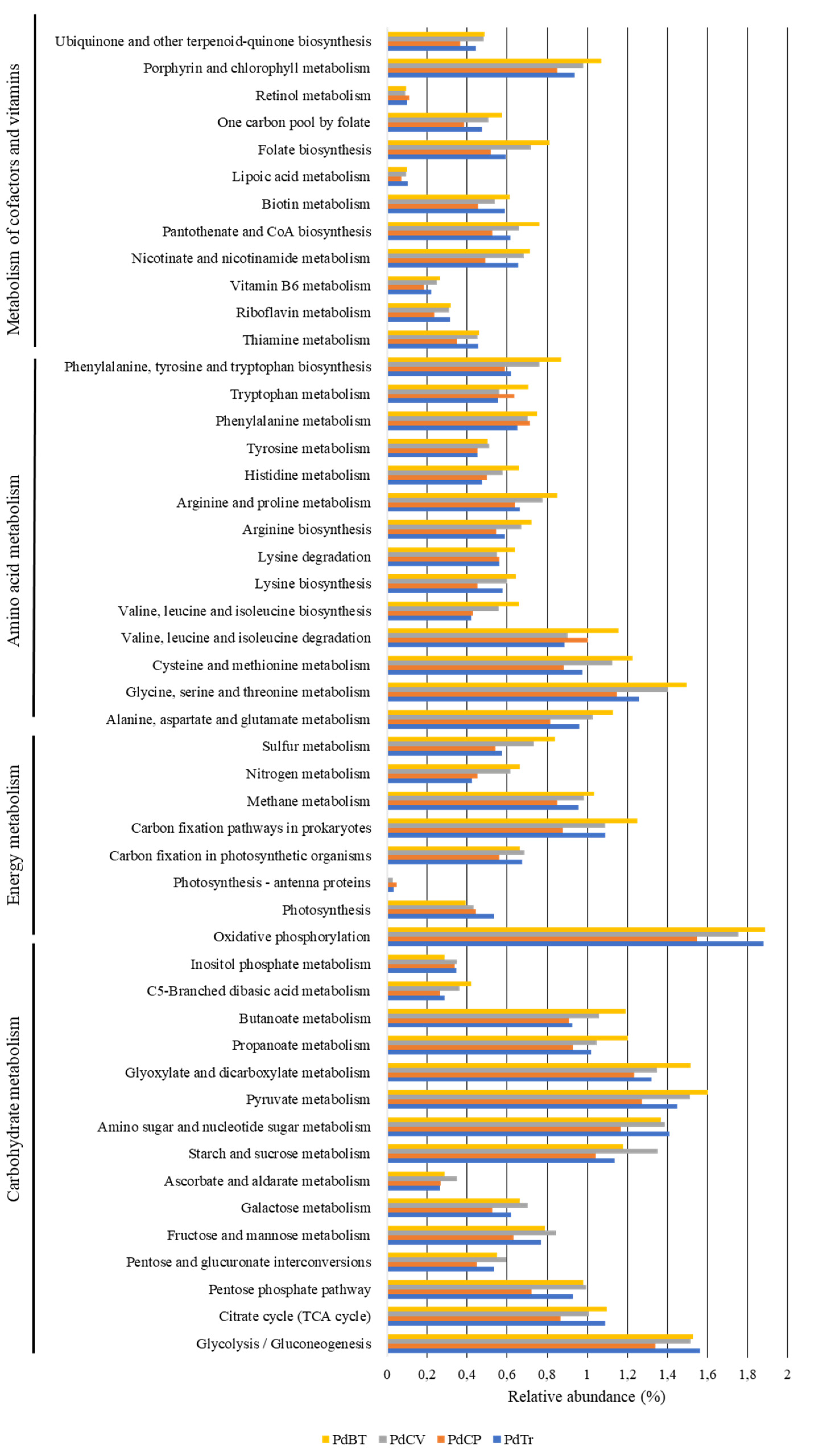
3.4. Prediction of Functional Diversity of the Gut Bacterial Communities
3.5. Prediction of LCB-Degrading Enzymes in the Bacterial Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kostanjšek, R.; Milatovib, M.; Ktrus, J. Endogenous origin of endo-β-1,4-glucanase in common woodlouse Porcellio scaber (Crustacea, Isopoda). J. Comp. Physiol. B 2010, 180, 1143–1153. [Google Scholar] [CrossRef]
- Bredon, M.; Dittmer, J.; Noël, C.; Moumen, B.; Bouchon, D. Lignocellulose degradation at the holobiont level: Teamwork in a keystone soil invertebrate. Microbiome 2018, 6, 162. [Google Scholar] [CrossRef] [Green Version]
- Bouchon, D.; Zimmer, M.; Dittmer, J. The terrestrial isopod microbiome: An all-in-one toolbox for animal-microbe interactions of ecological relevance. Front. Microbiol. 2016, 7, 1472. [Google Scholar] [CrossRef] [Green Version]
- de Gonzalo, G.; Colpa, D.I.; Habib, M.H.; Fraaije, M.W. Bacterial enzymes involved in lignin degradation. J. Biotechnol. 2016, 236, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Cragg, S.M.; Beckham, G.T.; Bruce, N.C.; Bugg, T.D.; Distel, D.L.; Dupree, P.; Etxabe, A.G.; Goodell, B.S.; Jellison, J.; McGeehan, J.E.; et al. Lignocellulose degradation mechanisms across the Tree of Life. Curr. Opin. Chem. Biol. 2015, 29, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Tolalpa, L.; Norder, J.; van Elsas, J.D.; Falcao Salles, J. Halotolerant microbial consortia able to degrade highly recalcitrant plant biomass substrate. Appl. Microbiol. Biotechnol. 2018, 102, 2913–2927. [Google Scholar] [CrossRef] [Green Version]
- Bashir, Z.; Kondapalli, V.K.; Adlakha, N.; Sharma, A.; Bhatnagar, R.K.; Chandel, G.; Yazdani, S.S. Diversity and functional significance of cellulolytic microbes living in termite, pill-bug and stem-borer guts. Sci. Rep. 2013, 3, 2558. [Google Scholar] [CrossRef] [Green Version]
- Mathews, S.L.; Pawlak, J.; Gruden, A.M. Bacterial biodegradation and bioconversion of industrial lignocellulosic streams. Appl. Microbiol. Biotechnol. 2015, 99, 2939–2954. [Google Scholar] [CrossRef]
- Janusz, G.; Pawlik, A.; Sulej, J.; Swiderska-Burek, U.; Jarosz-Wilkolazka, A.; Paszczynski, A. Lignin degradation: Microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiol. Rev. 2017, 41, 941–962. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, M.; Bharathiraja, C.; Pandiarajan, J.; Prasanna, V.A.; Rajendhran, J.; Gunasekaran, P. Insect gut microbiome—An unexploited reserve for biotechnological application. Asian Pac. J. Trop. Biomed. 2014, 4, 16–21. [Google Scholar] [CrossRef]
- Větrovský, T.; Steffen, K.T.; Baldrian, P. Potential of cometabolic transformation of polysaccharides and lignin in lignocellulose by soil Actinobacteria. PLoS ONE 2014, 9, e89108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulyshen, M.D. Wood decomposition as influenced by invertebrates. Biol. Rev. Camb. Philos. Soc. 2016, 91, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.T.; Speranza, M.; Ruiz-Dueñas, F.J.; Ferreira, P.; Camarero, S.; Guillén, F.; Martínez, M.J.; Gutiérrez, A.; del Río, J.C. Biodegradation of lignocellulosics: Microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 2005, 8, 195–204. [Google Scholar] [PubMed]
- Iyer, A.P.; Mahadevan, A. Lignin degradation by bacteria. Prog. Ind. Microbiol. 2002, 36, 311–330. [Google Scholar] [CrossRef]
- Caseiro, I.; Santos, S.; Sousa, J.P.; Nogueira, A.J.A.; Soares, A.M.V.M. Optimization of culture conditions of Porcellio dilatatus (Crustacea: Isopoda) for laboratory test development. Ecotoxicol. Environ. Saf. 2000, 47, 285–291. [Google Scholar] [CrossRef]
- Ribeiro, S.; Sousa, J.P.; Nogueira, A.J.; Soares, A.M. Effect of endosulfan and parathion on energy reserves and physiological parameters of the terrestrial isopod Porcellio dilatatus. Ecotoxicol. Environ. Saf. 2001, 49, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaux, R.; Pichon, S.; Hatira, H.B.; Doublet, V.; Grève, P.; Marcadé, I.; Braquart-Varnier, C.; Souty-Grosset, C.; Charfi-Cheikhrouha, F.; Bouchon, D. Widespread Wolbachia infection in terrestrial isopods and other crustaceans. ZooKeys 2012, 176, 123–131. [Google Scholar] [CrossRef]
- Ye, Y.H.; Seleznev, A.; Flores, H.A.; Woolfit, M.; McGraw, E.A. Gut microbiota in Drosophila melanogaster interacts with Wolbachia but does not contribute to Wolbachia-mediated antiviral protection. J. Invertebr. Pathol. 2016, 143, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, D.; Cordaux, R.; Grève, P. Rickettsiella, intracellular pathogens of arthropods. Manip. Tenants Bact. Assoc. Arthropods 2011, 2, 127. [Google Scholar]
- Wang, Y.; Stingl, U.; Anton-Erxleben, F.; Geisler, S.; Brune, A.; Zimmer, M. “Candidatus Hepatoplasma crinochetorum,” a new, stalk-forming lineage of mollicutes colonizing the midgut glands of a terrestrial isopod. Appl. Environ. Microbiol. 2004, 70, 6166–6172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostanjšek, R.; Strus, J.; Avgustin, G. “Candidatus Bacilloplasma,” a novel lineage of mollicutes associated with the hindgut wall of the terrestrial isopod Porcellio scaber (Crustacea: Isopoda). Appl. Environ. Microbiol. 2007, 73, 5566–5573. [Google Scholar] [CrossRef]
- Ellegaard, K.M.; Klasson, L.; Naslund, K.; Bourtzis, K.; Andersson, S.G. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 2013, 9, e1003381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef]
- Yun, J.H.; Roh, S.W.; Whon, T.W.; Jung, M.J.; Kim, M.S.; Park, D.S.; Yoon, C.; Nam, Y.D.; Kim, Y.J.; Choi, J.H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duron, O.; Bouchon, D.; Boutin, S.; Bellamy, L.; Zhou, L.; Engelstädter, J.; Hurst, G.D. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 2008, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Cordaux, R.; Paces-Fessy, M.; Raimond, M.; Michel-Salzat, A.; Zimmer, M.; Bouchon, D. Molecular characterization and evolution of arthropod-pathogenic Rickettsiella Bacteria. Appl. Environ. Microbiol. 2007, 73, 5045–5047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vago, C.; Meynadier, G.; Juchault, P.; Legrand, J.J.; Amargier, A.; Duthoit, L.A. Rickettsial disease of isopod crustaceans. C. R. Acad. Sci. 1970, 271, 2061–2063. [Google Scholar]
- Federici, B.A. Diseases of terrestrial isopods. Symp. Zool. Soc. Lond. 1984, 53, 233–245. [Google Scholar]
- Abd, E.M.; Holdich, D.M. The occurrence of a Rickettsial disease in British woodlice (Crustacea, Isopoda, Oniscidea) populations. J. Invertebr. Pathol. 1987, 49, 252–258. [Google Scholar] [CrossRef]
- Kleespies, R.G.; Federici, B.A.; Leclerque, A. Ultrastructural characterization and multilocus sequence analysis (MLSA) of “Candidatus Rickettsiella isopodorum”, a new lineage of intracellular bacteria infecting woodlice (Crustacea:Isopoda). Syst. Appl. Microbiol. 2014, 37, 351–359. [Google Scholar] [CrossRef]
- Delhoumi, M.; Zaabar, W.; Bouslama, M.F.; Achouri, M.S. Effect of symbiont acquisition on growth, survival and fertility of the terrestrial isopod Porcellionides pruinosus (Crustacea, Oniscidea). Invertebr. Reprod. Dev. 2019, 63, 60–66. [Google Scholar] [CrossRef]
- Fraune, S.; Zimmer, M. Host-specificity of environmentally transmitted Mycoplasma-like isopod symbionts. Environ. Microbiol. 2008, 10, 2497–2504. [Google Scholar] [CrossRef]
- Dittmer, J.; Bouchon, D. Feminizing Wolbachia influence microbiota composition in the terrestrial isopod Armadillidium vulgare. Sci. Rep. 2018, 8, 6998. [Google Scholar] [CrossRef]
- Bredon, M.; Herran, B.; Bertaux, J.; Grève, P.; Moumen, B.; Bouchon, D. Isopod holobionts as promising models for lignocellulose degradation. Biotechnol. Biofuels 2020, 13, 49. [Google Scholar] [CrossRef]
- Kostanjšek, R.; Strus, J.; Avgustin, G. Genetic diversity of bacteria associated with the hindgut of the terrestrial crustacean Porcellio scaber (Crustacea: Isopoda). FEMS Microbiol. 2002, 40, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J.; Lesobre, J.; Moumen, B.; Bouchon, D. Host origin and tissue microhabitat shaping the microbiota of the terrestrial isopod Armadillidium vulgare. FEMS Microbiol. 2016, 92, fiw063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madžarić, S.; Kos, M.; Drobne, D.; Hočevar, M.; Jemec Kokalj, A. Integration of behavioral tests and biochemical biomarkers of terrestrial isopod Porcellio scaber (Isopoda, Crustacea) is a promising methodology for testing environmental safety of chars. Environ. Pollut. 2018, 234, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Bredon, M.; Herran, B.; Lheraud, B.; Bertaux, J.; Grève, P.; Moumen, B.; Bouchon, D. Lignocellulose degradation in isopods: New insights into the adaptation to terrestrial life. BMC Genom. 2019, 20, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drobne, D.; Rupnik, M.; Lapanje, A.; Strus, J.; Janc, M. Isopod gut microflora parameters as endpoints in toxicity studies. Environ. Toxicol. Chem. 2002, 21, 604–609. [Google Scholar] [CrossRef]
- Vargas-Asensio, G.; Pinto-Tomas, A.; Rivera, B.; Hernandez, M.; Hernandez, C.; Soto-Montero, S.; Murillo, C.; Sherman, D.H.; Tamayo-Castillo, G. Uncovering the cultivable microbial diversity of costa rican beetles and its ability to break down plant cell wall components. PLoS ONE 2014, 9, e113303. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Opens external link in new window. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Opens external link in new window. Nucleic Acids Res. 2014, 42, 643–648. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data, 2020. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst—A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Dube, J.P.; Valverde, A.; Steyn, J.M.; Cowan, D.A.; van der Waals, J.E. Differences in bacterial diversity, composition and function due to long-term agriculture in soils in the eastern free state of south Africa. Diversity 2019, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Horváthová, T.; Babik, W.; Bauchinger, U. Biofilm feeding: Microbial colonization of food promotes the growth of a detritivorous arthropod. ZooKeys 2016, 577, 25–41. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Yang, L.; Huang, S.; Su, X.; Li, Y.; Wang, F.; Wang, E.; Kang, N.; Xu, J.; Song, A. Comparative Gut Microbiomes of Four Species Representing the Higher and the Lower Termites. J. Insect Sci. 2016, 16, 97. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Lian, B.; Wu, C.; Guo, P. A comparative study of gut microbiota profiles of earthworms fed in three different substrates. Symbiosis 2018, 74, 21–29. [Google Scholar] [CrossRef]
- Berlanga, M.; Lorens, C.; Comas, J.; Guerrero, R. Gut Bacterial Community of the Xylophagous Cockroaches Cryptocercus punctulatus and Parasphaeria boleiriana. PLoS ONE 2017, 11, e152400. [Google Scholar] [CrossRef]
- Correia, D.S.; Passos, S.R.; Proença, D.N.; Morais, P.V.; Xavier, G.R.; Correia, M.E. Microbial diversity associated to the intestinal tract of soil invertebrates. Appl. Soil. Ecol. 2018, 131, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Bolaños, L.M.; Rosenblueth, M.; Castillo-Ramírez, S.; Figuier-Huttin, G.; Martínez-Romero, E. Species-specific diversity of novel bacterial lineages and differential abundance of predicted pathways for toxic compound degradation in scorpion gut microbiota. Environ. Microbiol. 2016, 18, 1364–1378. [Google Scholar] [CrossRef]
- Benjamino, J.; Graf, J. Characterization of the Core and Caste-Specific Microbiota in the Termite, Reticulitermes flavipes. Front. Microbiol. 2016, 7, 171. [Google Scholar] [CrossRef] [Green Version]
- Abdelrhman, K.; Bacci, G.; Marras, B.; Nistri, A.; Schintu, M.; Ugolini, A.; Mengoni, A. Exploring the bacterial gut microbiota of supralittoral talitrid amphipods. Microbiol. Res. 2017, 168, 74–84. [Google Scholar] [CrossRef]
- Kolasa, M.; Ścibior, R.; Mazur, M.A.; Kubisz, D.; Dudek, K.; Kajtoch, Ł. How hosts taxonomy, trophy, and endosymbionts shape microbiome diversity in beetles. Microb. Ecol. 2019, 78, 995–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, G.; Eunen, V.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrndorff, S.; de Jonge, N.; Kjerulf Hansen, J.K.; Lauritzen, J.M.S.; Spanggaard, L.H.; Sørensen, M.H.; Yde, M.; Nielsen, J.L. Diversity and metabolic potential of the microbiota associated with a soil arthropod. Sci. Rep. 2018, 8, 2491. [Google Scholar] [CrossRef] [Green Version]
- Lardies, M.A.; Carter, M.J.; Bozinovic, F. Dietary effects on life history traits in a terrestrial isopod: The importance of evaluating maternal effects and trade-offs. Oecologia 2004, 138, 387–395. [Google Scholar] [CrossRef]
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009, 23, 38–47. [Google Scholar] [CrossRef]
- Hmelo, L.R. Quorum Sensing in Marine Microbial Environments. Ann. Rev. Mar. Sci. 2017, 9, 257–281. [Google Scholar] [CrossRef]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin Vi, R.L.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [Green Version]
- Horváthová, T.; Bauchinger, U. Biofilm improves isopod growth independent of the dietary cellulose. Physiol. Biochem. Zool. 2019, 92, 6. [Google Scholar] [CrossRef] [PubMed]
- Delhoumi, M.; Catania, V.; Zaabar, W.; Tolone, M.; Quatrini, P.; Achouri, M.S. The gut microbiota structure of the terrestrial isopod Porcellionides pruinosus (Isopoda: Oniscidea). Eur. Zool. J. 2020, 87, 357–368. [Google Scholar] [CrossRef]
- Bredon, M.; Depuydt, E.; Brisson, L.; Moulin, L.; Charles, C.; Haenn, S.; Moumen, B.; Bouchon, D. Effects of dysbiosis and dietary manipulation on the digestive microbiota of a detritivorous arthropod. Microorganisms 2021, 11, 148. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, C.; Tiago, I.; Veríssimo, A. Guts Bacterial Communities of Porcellio dilatatus: Symbionts Predominance, Functional Significance and Putative Biotechnological Potential. Microorganisms 2022, 10, 2230. https://doi.org/10.3390/microorganisms10112230
Coelho C, Tiago I, Veríssimo A. Guts Bacterial Communities of Porcellio dilatatus: Symbionts Predominance, Functional Significance and Putative Biotechnological Potential. Microorganisms. 2022; 10(11):2230. https://doi.org/10.3390/microorganisms10112230
Chicago/Turabian StyleCoelho, Catarina, Igor Tiago, and António Veríssimo. 2022. "Guts Bacterial Communities of Porcellio dilatatus: Symbionts Predominance, Functional Significance and Putative Biotechnological Potential" Microorganisms 10, no. 11: 2230. https://doi.org/10.3390/microorganisms10112230