
A Methylotrophic Bacterium Growing with the Antidiabetic Drug Metformin as Its Sole Carbon, Nitrogen and Energy Source
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Characterisation of Strain MD1
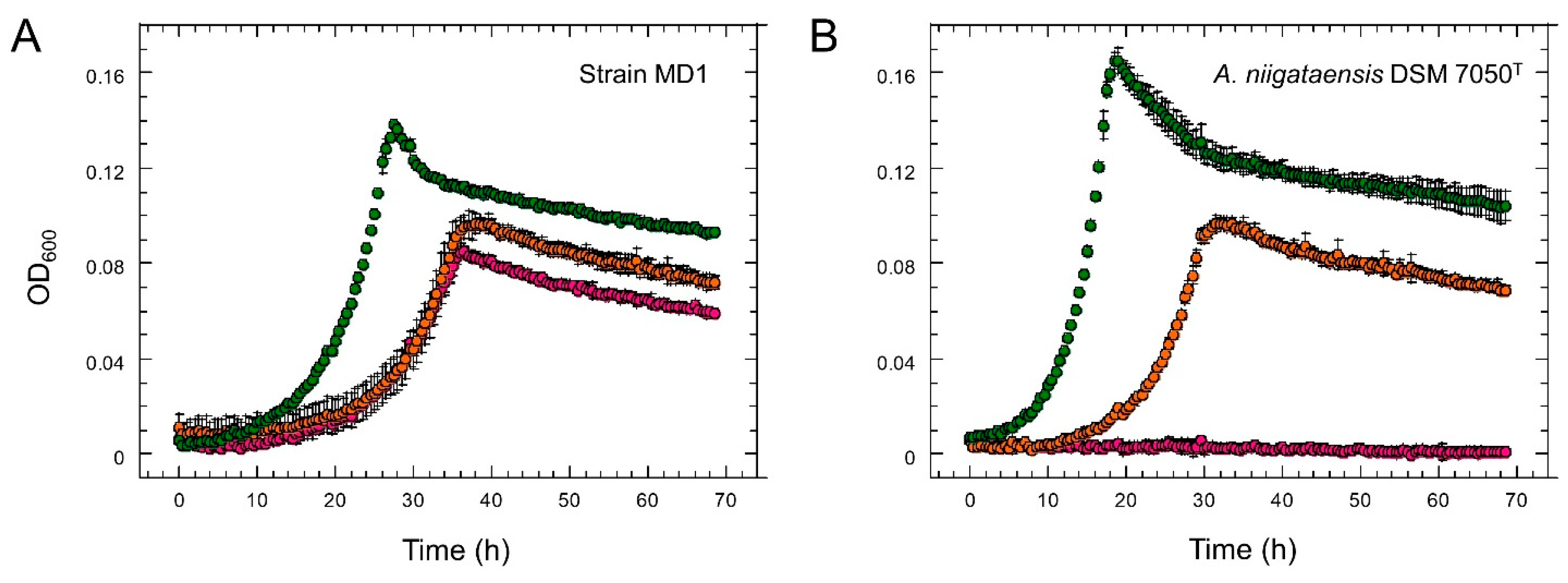
2.2. Growth Experiments
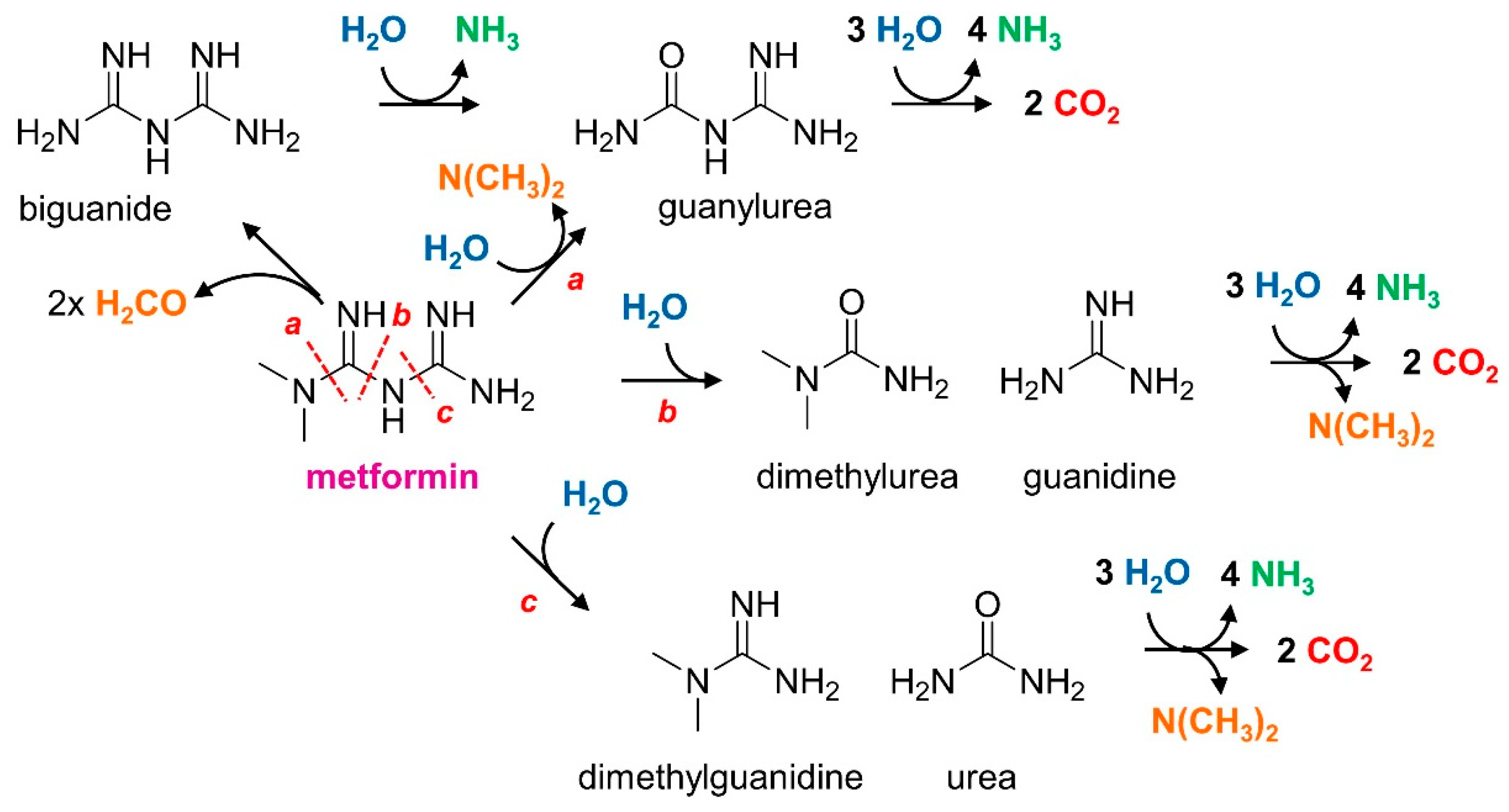
2.3. Analysis of Metformin and Transformation Products
2.4. Genome Sequencing, Assembly, Annotation and Analysis
2.5. Proteomic Analysis
2.6. RNA-Seq Analysis
2.7. Mini-Transposon Mutagenesis
2.8. Data
3. Results
3.1. Isolation and Characterization of Strain MD1
3.2. Genome Analysis
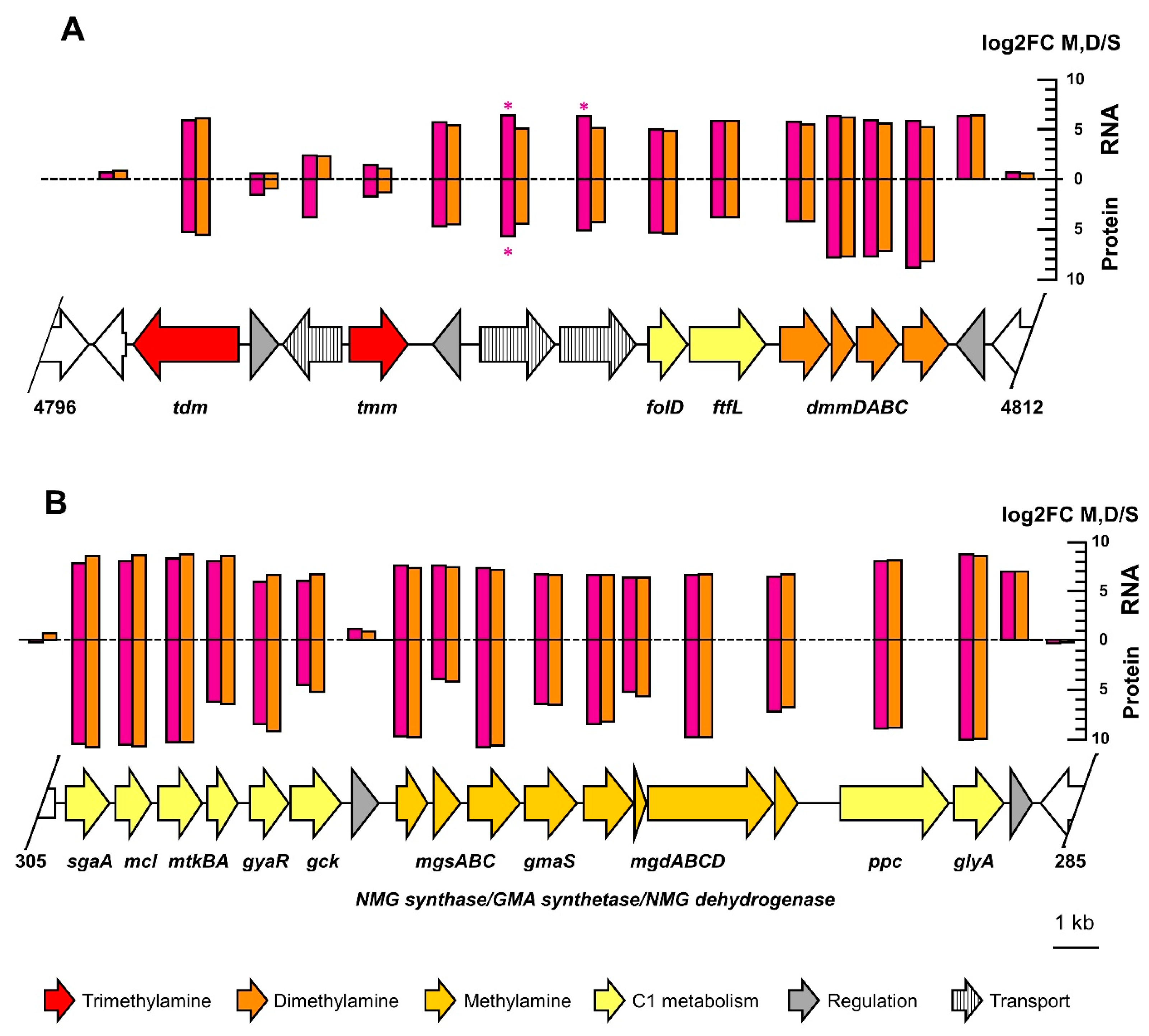
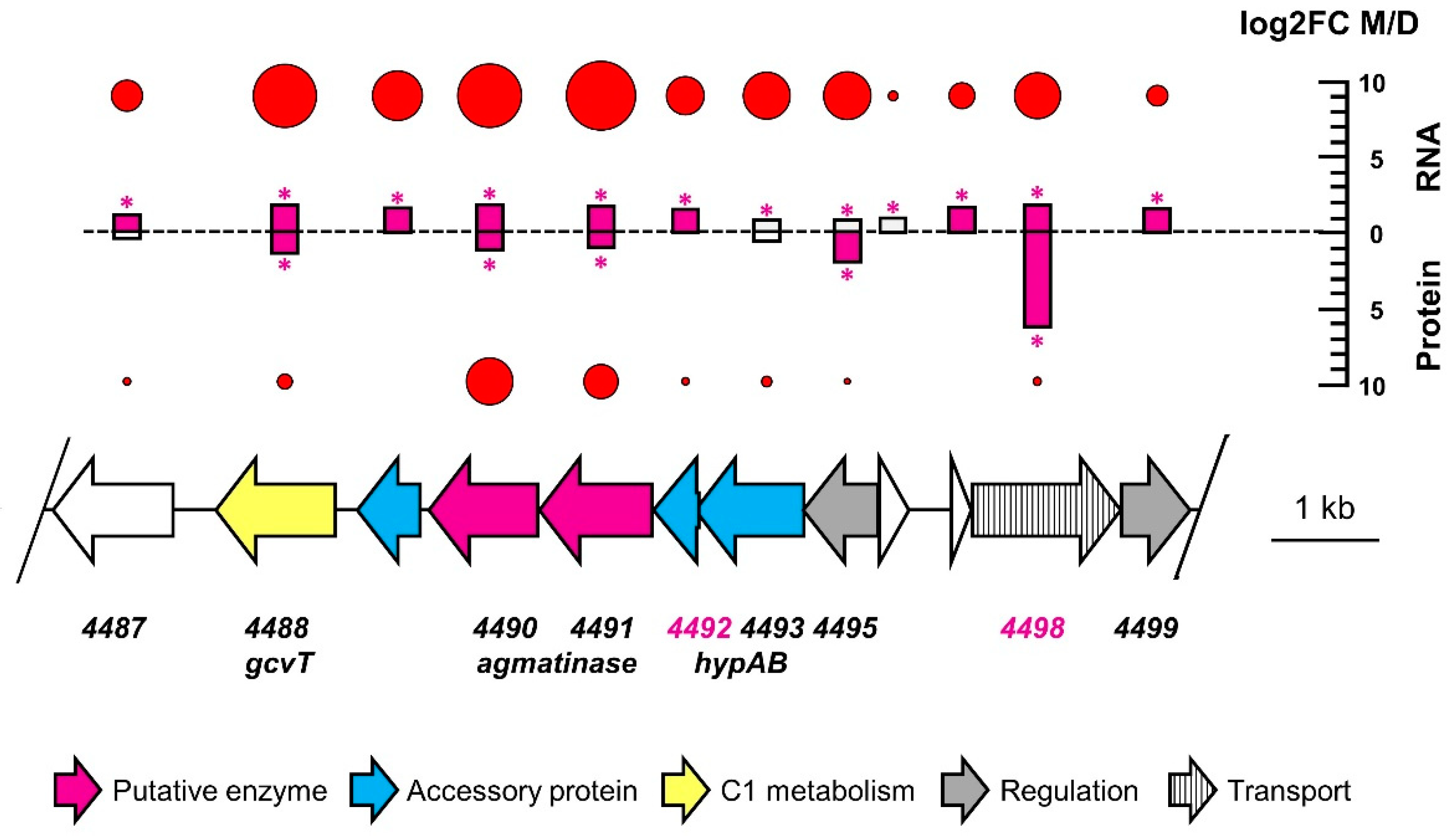
3.3. Comparative Proteomics and Transcriptomics
3.4. Mini-Transposon Mutagenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patel, M.; Kumar, R.; Kishor, K.; Mlsna, T.; Pittman, C.U.; Mohan, D. Pharmaceuticals of emerging concern in aquatic systems: Chemistry, occurrence, effects, and removal Methods. Chem. Rev. 2019, 119, 3510–3673. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, J.L.; Boxall, A.B.A.; Kolpin, D.W.; Leung, K.M.Y.; Lai, R.W.S.; Galbán-Malagón, C.; Adell, A.D.; Mondon, J.; Metian, M.; Marchant, R.A.; et al. Pharmaceutical pollution of the world’s rivers. Proc. Natl. Acad. Sci. USA 2022, 119, e2113947119. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.; Carney Almroth, B.M.; Collins, C.D.; Cornell, S.; de Wit, C.A.; Diamond, M.L.; Fantke, P.; Hassellöv, M.; MacLeod, M.; Ryberg, M.W.; et al. Outside the safe operating space of the planetary boundary for novel entities. Environ. Sci. Technol. 2022, 56, 1510–1521. [Google Scholar] [CrossRef]
- Wackett, L.P.; Robinson, S.L. The ever-expanding limits of enzyme catalysis and biodegradation: Polyaromatic, polychlorinated, polyfluorinated, and polymeric compounds. Biochem. J. 2020, 477, 2875–2891. [Google Scholar] [CrossRef]
- Poursat, B.A.J.; van Spanning, R.J.M.; Braster, M.; Helmus, R.; de Voogt, P.; Parsons, J.R. Biodegradation of metformin and its transformation product, guanylurea, by natural and exposed microbial communities. Ecotoxicol. Environ. Safety 2019, 182, 109414. [Google Scholar] [CrossRef]
- Qi, M.Y.; Liang, B.; Zhang, L.; Ma, X.D.; Yan, L.; Dong, W.C.; Kong, D.Y.; Zhang, L.Y.; Zhu, H.Z.; Gao, S.H.; et al. Microbial interactions drive the complete catabolism of the antibiotic sulfamethoxazole in activated sludge microbiomes. Environ. Sci. Technol. 2021, 55, 3270–3282. [Google Scholar] [CrossRef]
- Rios-Miguel, A.B.; Smith, G.J.; Cremers, G.; van Alen, T.; Jetten, M.S.M.; Camp, H.; Welte, C.U. Microbial paracetamol degradation involves a high diversity of novel amidase enzyme candidates. Water Res. X 2022, 16, 100152. [Google Scholar] [CrossRef]
- Malhotra, H.; Kaur, S.; Phale, P.S. Conserved metabolic and evolutionary themes in microbial degradation of carbamate pesticides. Front. Microbiol. 2021, 12, 648868. [Google Scholar] [CrossRef]
- Chistoserdova, L. Methylotrophs and Methylotroph Communities; Caister Academic Press: Poole, UK, 2019; p. 272. [Google Scholar] [CrossRef]
- Evans, P.N.; Boyd, J.A.; Leu, A.O.; Woodcroft, B.; Parks, D.H.; Hugenholtz, P.; Tyson, G.W. An evolving view of methane metabolism in the Archaea. Nature Rev. Microbiol. 2019, 17, 219–232. [Google Scholar] [CrossRef]
- Romero, R.; Erez, O.; Huttemann, M.; Huttemann, M.; Panaitescu, B.; Conde-Agudelo, A.; Pacora, P.; Yoon, B.H.; Grossman, L.I. Metformin, the aspirin of the 21st century: Its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity. Amer. J. Obstet. Gynecol. 2017, 217, 282–302. [Google Scholar] [CrossRef]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is it a drug for all reasons and diseases? Metabol. Clin. Exp. 2022, 133, 155223. [Google Scholar] [CrossRef] [PubMed]
- Di Magno, L.; Di Pastena, F.; Bordone, R.; Coni, S.; Canettieri, G. The mechanism of action of biguanides: New answers to a complex question. Cancers 2022, 14, 3220. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio-Albuquerque, E.P.; Cusioli, L.F.; Bergamasco, R.; Gigliolli, A.A.S.; Lupepsa, L.; Paupitz, B.R.; Barbieri, P.A.; Borin-Carvalho, L.A.; Portela-Castro, A.L.D. Metformin environmental exposure: A systematic review. Environ. Toxicol. Pharmacol. 2021, 83, 103588. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Sillanpaa, M.; Jacob, M.M.; Vo, D.V.N. Metformin as an emerging concern in wastewater: Occurrence, analysis and treatment methods. Environ. Res. 2022, 213, 113613. [Google Scholar] [CrossRef]
- He, Y.Z.; Zhang, Y.Y.; Ju, F. Metformin contamination in global waters: Biotic and abiotic transformation, byproduct generation and toxicity, and evaluation as a pharmaceutical indicator. Environ. Sci. Technol. 2022, 56, 13528–13545. [Google Scholar] [CrossRef]
- Nielsen, K.M.; DeCamp, L.; Birgisson, M.; Palace, V.P.; Kidd, K.A.; Parrott, J.L.; McMaster, M.E.; Alaee, M.; Blandford, N.; Ussery, E.J. Comparative effects of embryonic metformin exposure on wild and laboratory-spawned fathead minnow (Pimephales promelas) populations. Environ. Sci. Technol. 2022, 56, 10193–10203. [Google Scholar] [CrossRef]
- Wensink, M.J.; Lu, Y.; Tian, L.; Shaw, G.M.; Rizzi, S.; Jensen, T.K.; Mathiesen, E.R.; Skakkebaek, N.E.; Lindahl-Jacobsen, R.; Eisenberg, M.L. Preconception antidiabetic drugs in men and birth defects in offspring: A nationwide cohort study. Annals Intern. Med. 2022, 175, 665–673. [Google Scholar] [CrossRef]
- Caldwell, D.J.; D’Aco, V.; Davidson, T.; Kappler, K.; Murray-Smith, R.J.; Owen, S.F.; Robinson, P.F.; Simon-Hettich, B.; Straub, J.O.; Tell, J. Environmental risk assessment of metformin and its transformation product guanylurea: II. Occurrence in surface waters of Europe and the United States and derivation of predicted no-effect concentrations. Chemosphere 2019, 216, 855–865. [Google Scholar] [CrossRef]
- Trautwein, C.; Berset, J.D.; Wolschke, H.; Kümmerer, K. Occurrence of the antidiabetic drug metformin and its ultimate transformation product guanylurea in several compartments of the aquatic cycle. Environ. Internat. 2014, 70, 203–212. [Google Scholar] [CrossRef]
- Trautwein, C.; Kummerer, K. Incomplete aerobic degradation of the antidiabetic drug metformin and identification of the bacterial dead-end transformation product guanylurea. Chemosphere 2011, 85, 765–773. [Google Scholar] [CrossRef]
- Straub, J.O.; Caldwell, D.J.; Davidson, T.; D’Aco, V.; Kappler, K.; Robinson, P.F.; Simon-Hettich, B.; Tell, J. Environmental risk assessment of metformin and its transformation product guanylurea. I. Environmental fate. Chemosphere 2019, 216, 844–854. [Google Scholar] [CrossRef]
- Tisler, S.; Zwiener, C. Aerobic and anaerobic formation and biodegradation of guanyl urea and other transformation products of metformin. Water Res. 2019, 149, 130–135. [Google Scholar] [CrossRef]
- Scheurer, M.; Michel, A.; Brauch, H.J.; Ruck, W.; Sacher, F. Occurrence and fate of the antidiabetic drug metformin and its metabolite guanylurea in the environment and during drinking water treatment. Water Res. 2012, 46, 4790–4802. [Google Scholar] [CrossRef]
- Schneider, N.O.; Tassoulas, L.J.; Zeng, D.Y.; Laseke, A.J.; Reiter, N.J.; Wackett, L.P.; St Maurice, M. Solving the conundrum: Widespread proteins annotated for urea metabolism in bacteria are carboxyguanidine deiminases mediating nitrogen assimilation from guanidine. Biochemistry 2020, 59, 3258–3270. [Google Scholar] [CrossRef]
- Tassoulas, L.J.; Robinson, A.; Martinez-Vaz, B.; Aukema, K.G.; Wackett, L.P. Filling in the gaps in metformin biodegradation: A new enzyme and a metabolic pathway for guanylurea. Appl. Environ. Microbiol. 2021, 87, e03003–e03020. [Google Scholar] [CrossRef]
- Hillmann, K.B.; Niehaus, T.D. Genome sequences of two Pseudomonas isolates that can use metformin as the sole nitrogen source. Microbiol. Resour. Announc. 2022, 11, e00639-22. [Google Scholar] [CrossRef]
- Gruffaz, C.; Muller, E.E.L.; Louhichi-Jelail, Y.; Nelli, Y.R.; Guichard, G.; Bringel, F. Genes of the N-methylglutamate pathway are essential for growth of Methylobacterium extorquens DM4 with monomethylamine. Appl. Environ. Microbiol. 2014, 80, 3541–3550. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, J.R.; Sato, T.; Weightman, A.J.; Martin, T.A.; Fry, J.C.; Hiom, S.J.; Wade, W.G. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 1998, 64, 795–799. [Google Scholar] [CrossRef] [Green Version]
- Sjoholm, O.R.; Aamand, J.; Sorensen, J.; Nybroe, O. Degrader density determines spatial variability of 2,6-dichlorobenzamide mineralisation in soil. Environ. Pollut. 2010, 158, 292–298. [Google Scholar] [CrossRef]
- Suzuki, M.T.; Taylor, L.T.; DeLong, E.F. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl. Environ. Microbiol. 2000, 66, 4605–4614. [Google Scholar] [CrossRef] [Green Version]
- Cashion, P.; Holder-Franklin, M.A.; McCully, J.; Franklin, M. A rapid method for the base ratio determination of bacterial DNA. Anal. Biochem. 1977, 81, 461–466. [Google Scholar] [CrossRef]
- De Ley, J.; Cattoir, H.; Reynaerts, A. The quantitative measurement of DNA hybridization from renaturation rates. Eur. J. Biochem. 1970, 12, 133–142. [Google Scholar] [CrossRef]
- Huss, V.A.; Festl, H.; Schleifer, K.H. Studies on the spectrophotometric determination of DNA hybridization from renaturation rates. Syst. Appl. Microbiol. 1983, 4, 184–192. [Google Scholar] [CrossRef]
- Bai, Y.; Muller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Munch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Horemans, B.; Lood, C.; T’Syen, J.; van Noort, V.; Lavigne, R.; Ellegaard-Jensen, L.; Hylling, O.; Aamand, J.; Springael, D.; et al. The complete genome of 2,6-dichlorobenzamide (BAM) degrader Aminobacter sp. MSH1 suggests a polyploid chromosome, phylogenetic reassignment, and functions of plasmids. Sci. Rep. 2021, 11, 18943. [Google Scholar] [CrossRef]
- Deitch, A.D. An improved Sakaguchi reaction for microspectrophotometric use. J. Histochem. Cytochem. 1961, 9, 477–483. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 5 September 2022).
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Laboratory: Berkeley, CA USA, 2014. LBNL Report LBNL-7065E. Available online: https://escholarship.org/uc/item/1h3515gn (accessed on 30 September 2022).
- Boetzer, M.; Pirovano, W. SSPACE-LongRead: Scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform. 2014, 15, 211. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Vallenet, D.; Calteau, A.; Dubois, M.; Amours, P.; Bazin, A.; Beuvin, M.; Burlot, L.; Bussell, X.; Fouteau, S.; Gautreau, G.; et al. MicroScope: An integrated platform for the annotation and exploration of microbial gene functions through genomic, pangenomic and metabolic comparative analysis. Nucl. Acids Res. 2020, 48, D579–D589. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucl. Acids Res. 2021, 50, D801–D807. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Roche, B.; Garcia-Rivera, M.A.; Normant, V.; Kuhn, L.; Hammann, P.; Bronstrup, M.; Mislin, G.L.A.; Schalk, I.J. A role for PchHI as the ABC transporter in iron acquisition by the siderophore pyochelin in Pseudomonas aeruginosa. Environ. Microbiol. 2022, 24, 866–877. [Google Scholar] [CrossRef]
- Bouyssie, D.; Hesse, A.M.; Mouton-Barbosa, E.; Rompais, M.; Macron, C.; Carapito, C.; de Peredo, A.G.; Coute, Y.; Dupierris, V.; Burel, A.; et al. Proline: An efficient and user-friendly software suite for large-scale proteomics. Bioinformatics 2020, 36, 3148–3155. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-f.; Romero A, D.; Guan, S.; Mamanova, L.; McDowall, K.J. A combination of improved differential and global RNA-seq reveals pervasive transcription initiation and events in all stages of the life-cycle of functional RNAs in Propionibacterium acnes, a major contributor to wide-spread human disease. BMC Genom. 2013, 14, 620. [Google Scholar] [CrossRef] [Green Version]
- Muller, E.; Hourcade, E.; Louhichi, Y.; Hammann, P.; Vuilleumier, S.; Bringel, F. Functional genomics of dichloromethane utilisation in Methylobacterium extorquens DM4. Environ. Microbiol. 2011, 13, 2518–2534. [Google Scholar] [CrossRef]
- Artuso, I.; Turrini, P.; Pirolo, M.; Lugli, G.A.; Ventura, M.; Visca, P. Phylogenomic reconstruction and metabolic potential of the genus Aminobacter. Microorganisms 2021, 9, 1332. [Google Scholar] [CrossRef]
- Urakami, T.; Araki, H.; Oyanagi, H.; Suzuki, K.I.; Komagata, K. Transfer of Pseudomonas aminovorans (den Dooren deJong 1926) to Aminobacter gen. nov. as Aminobacter aminovorans comb. nov. and description of Aminobacter aganoensis sp. nov. and Aminobacter niigataensis sp. nov. Int. J. Syst. Bacteriol. 1992, 42, 84–92. [Google Scholar] [CrossRef]
- Artuso, I.; Turrini, P.; Pirolo, M.; Lucidi, M.; Tescari, M.; Visaggio, D.; Mansi, A.; Lugli, G.A.; Ventura, M.; Visca, P. Phylogenomic analysis and characterization of carbon monoxide utilization genes in the family Phyllobacteriaceae with reclassification of Aminobacter carboxidus (Meyer et al. 1993, Hrdt et al. 2020) as Aminobacter lissarensis comb. nov. (McDonald et al. 2005). Syst. Appl. Microbiol. 2021, 44, 126199. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucl. Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucl. Acids Res. 2020, 49, D344–D354. [Google Scholar] [CrossRef]
- Czarnecki, J.; Dziewit, L.; Puzyna, M.; Prochwicz, E.; Tudek, A.; Wibberg, D.; Schluter, A.; Puhler, A.; Bartosik, D. Lifestyle-determining extrachromosomal replicon pAMV1 and its contribution to the carbon metabolism of the methylotrophic bacterium Paracoccus aminovorans JCM 7685. Environ. Microbiol. 2017, 19, 4536–4550. [Google Scholar] [CrossRef]
- Zhu, Y.; Jameson, E.; Parslow, R.A.; Lidbury, I.; Fu, T.; Dafforn, T.R.; Schäfer, H.; Chen, Y. Identification and characterization of trimethylamine N-oxide (TMAO) demethylase and TMAO permease in Methylocella silvestris BL2. Environ. Microbiol. 2014, 16, 3318–3330. [Google Scholar] [CrossRef] [Green Version]
- Latypova, E.; Yang, S.; Wang, Y.S.; Wang, T.S.; Chavkin, T.A.; Hackett, M.; Schäfer, H.; Kalyuzhnaya, M.G. Genetics of the glutamate-mediated methylamine utilization pathway in the facultative methylotrophic beta-proteobacterium Methyloversatilis universalis FAM5. Molec. Microbiol. 2010, 75, 426–439. [Google Scholar] [CrossRef]
- Yukl, E.T.; Davidson, V.L. Diversity of structures, catalytic mechanisms and processes of cofactor biosynthesis of tryptophylquinone-bearing enzymes. Arch. Biochem. Biophys. 2018, 654, 40–46. [Google Scholar] [CrossRef]
- Vannelli, T.; Messmer, M.; Studer, A.; Vuilleumier, S.; Leisinger, T. A corrinoid-dependent catabolic pathway for growth of a Methylobacterium strain with chloromethane. Proc. Natl. Acad. Sci. USA 1999, 96, 4615–4620. [Google Scholar] [CrossRef]
- Suarez, A.; Güttler, A.; Strätz, M.; Staendner, L.H.; Timmis, K.N.; Guzmán, C.A. Green fluorescent protein-based reporter systems for genetic analysis of bacteria including monocopy applications. Gene 1997, 196, 69–74. [Google Scholar] [CrossRef]
- Maturana, P.; Orellana, M.S.; Herrera, S.M.; Martinez, I.; Figueroa, M.; Martinez-Oyanedel, J.; Castro-Fernandez, V.; Uribe, E. Crystal structure of Escherichia coli agmatinase: Catalytic mechanism and residues relevant for substrate specificity. Int. J. Molec. Sci. 2021, 22, 4769. [Google Scholar] [CrossRef]
- Funck, D.; Sinn, M.; Fleming, J.R.; Stanoppi, M.; Dietrich, J.; Lopez-Igual, R.; Mayans, O.; Hartig, J.S. Discovery of a Ni2+-dependent guanidine hydrolase in bacteria. Nature 2022, 603, 515–523. [Google Scholar] [CrossRef]
- Wang, B.; Xu, Y.; Wang, X.; Yuan, J.S.; Johnson, C.H.; Young, J.D.; Yu, J.P. A guanidine-degrading enzyme controls genomic stability of ethylene-producing cyanobacteria. Nat. Commun. 2021, 12, 5150. [Google Scholar] [CrossRef]
- Hatton, C.E.; Brotherton, D.H.; Spencer, M.; Cameron, A.D. Structure of cytosine transport protein CodB provides insight into nucleobase-cation symporter 1 mechanism. EMBO J. 2022, 41, e110527. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Chromosome | Plasmid paAMD1 | Plasmid pbAMD1 | Plasmid pcAMD1 |
|---|---|---|---|---|
| Coverage (-fold) | 69.6 | 62.2 | 60.9 | 50.6 |
| Topology | circular | circular | circular | circular |
| Size (kb) | 4,947,010 | 363,670 | 24,942 | 21,575 |
| GC (%) | 63.5 | 63.3 | 62.9 | 59.6 |
| rRNA operons | 2 | - | - | - |
| tRNA | 50 | - | - | - |
| CDS | 5140 | 329 | 34 | 24 |
| Coding density (%) | 90.0 | 91.5 | 73.6 | 78.2 |
| Completeness/contamination 1 (%) | 99.3/0.4 | NA 2 | NA 2 | NA 2 |
| Repeat regions 3 (%) | 4.8 | 0.9 | 24.0 | 0 |
| CDS with EGGNOG annotation (%) | 86.7 | 94.6 | 64.7 | 62.5 |
| CDS with hydrolase prediction 4 | 599 | 59 | 3 | 3 |
| CDS, specific hydrolase prediction 5,6 | 81 | 13 | - | - |
| CDS, carboxylase prediction 6 | 42 | 2 | - | - |
| Detected | M ≠ S 2 | M > S 3 | D ≠ S 2 | D > S 3 | M/D ≠ S 2 | M/D > S 4 | M ≠ D/S 2 | M > D/S 5 | |
|---|---|---|---|---|---|---|---|---|---|
| RNA-Seq | 5509 | 1189 | 113 (2.1) | 1955 | 293 (5.3) | 749 | 105 (1.9) | 589 | 9 (0.2) |
| Proteomics | 1581 | 125 | 52 (3.3) | 194 | 58 (3.7) | 83 | 39 (2.5) | 22 | 6 (0.4) |
| Common | 1580 | 90 | 43 | 158 | 52 | 70 | 37 | 16 | 6 |
| Gene | Transcriptomics | Proteomics | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Conser- | Cluster | log2FC 1 | Reads | log2FC 1 | SC | ||||
| CDS | Annotation | vation 2 | Location | M vs. S | M vs. D | (M) 3 | M vs. S | M vs. D | (M) 4 |
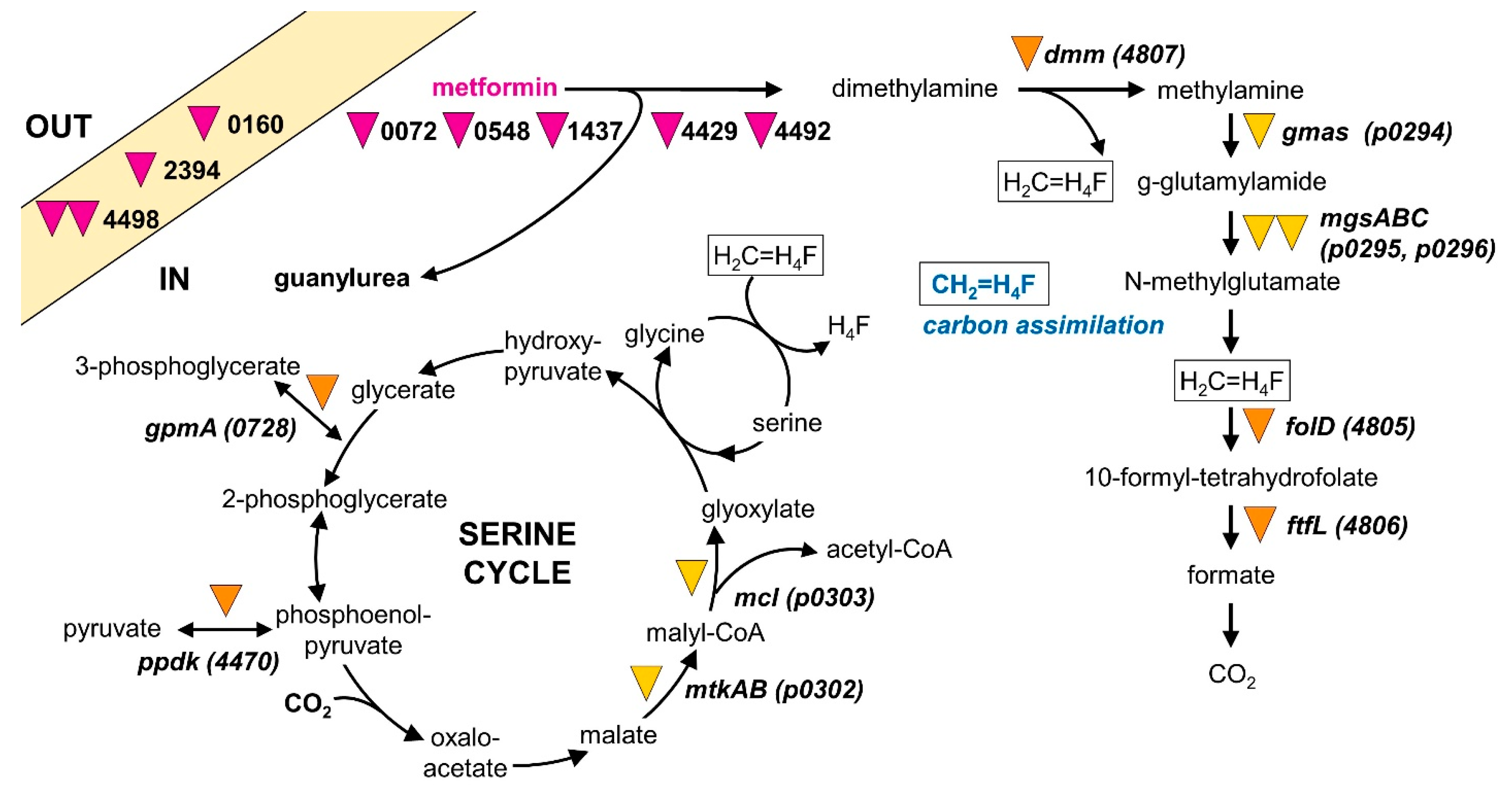
| 0072 | amidohydrolase | (no) 5 | 2/2 | 0.12 | 0.10 | 279 | - 6 | - | ND 7 |
| 0160 | ABC transporter | yes | 22/31 | −0.20 | - | 762 | - | - | ND 7 |
| 0548 | hydantoinase | no | 1/2 | 0.15 | 0.37 | 1364 | - | - | ND 7 |
| 1437 | diguanylate cyclase | yes | 2/2 | −0.34 | 0.97 | 1020 | - | - | ND 7 |
| 2394 | transporter, DUF486 | yes | 1/3 | −0.15 | −0.51 | 308 | - | - | ND 7 |
| 4429 | thiolase | yes | 1/4 | 0.03 | 0.03 | 1761 | −0.06 | 0.58 | 5 |
| 4492 | nickel incorporation | yes | 2/23 | 0.10 | 1.48 | 19,236 | −0.13 | −0.03 | 1 |
| 4498 | NCS transporter | yes | 2/3 | 0.17 | 1.78 | 28,303 | −0.91 | 6.22 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaignaud, P.; Gruffaz, C.; Borreca, A.; Fouteau, S.; Kuhn, L.; Masbou, J.; Rouy, Z.; Hammann, P.; Imfeld, G.; Roche, D.; et al. A Methylotrophic Bacterium Growing with the Antidiabetic Drug Metformin as Its Sole Carbon, Nitrogen and Energy Source. Microorganisms 2022, 10, 2302. https://doi.org/10.3390/microorganisms10112302
Chaignaud P, Gruffaz C, Borreca A, Fouteau S, Kuhn L, Masbou J, Rouy Z, Hammann P, Imfeld G, Roche D, et al. A Methylotrophic Bacterium Growing with the Antidiabetic Drug Metformin as Its Sole Carbon, Nitrogen and Energy Source. Microorganisms. 2022; 10(11):2302. https://doi.org/10.3390/microorganisms10112302
Chicago/Turabian StyleChaignaud, Pauline, Christelle Gruffaz, Adrien Borreca, Stéphanie Fouteau, Lauriane Kuhn, Jérémy Masbou, Zoé Rouy, Philippe Hammann, Gwenaël Imfeld, David Roche, and et al. 2022. "A Methylotrophic Bacterium Growing with the Antidiabetic Drug Metformin as Its Sole Carbon, Nitrogen and Energy Source" Microorganisms 10, no. 11: 2302. https://doi.org/10.3390/microorganisms10112302