
Ca2+/Calmodulin-Dependent Protein Kinase II Inhibits Hepatitis B Virus Replication from cccDNA via AMPK Activation and AKT/mTOR Suppression

 , , , , , , ,
, , , , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Cell Culture
2.3. Transfection
2.4. Establishment of Stable Cells
2.5. SDS-PAGE and Western Blotting
2.6. Core Particle Immunoblotting
2.7. Northern and Southern Blotting
2.8. HBV Preparation and Infection
2.9. cccDNA Extraction
2.10. Drug Treatment
2.11. Luciferase Reporter Assay
2.12. Quantitative Real-Time RT-PCR and PCR
2.13. Statistical Analysis
3. Results
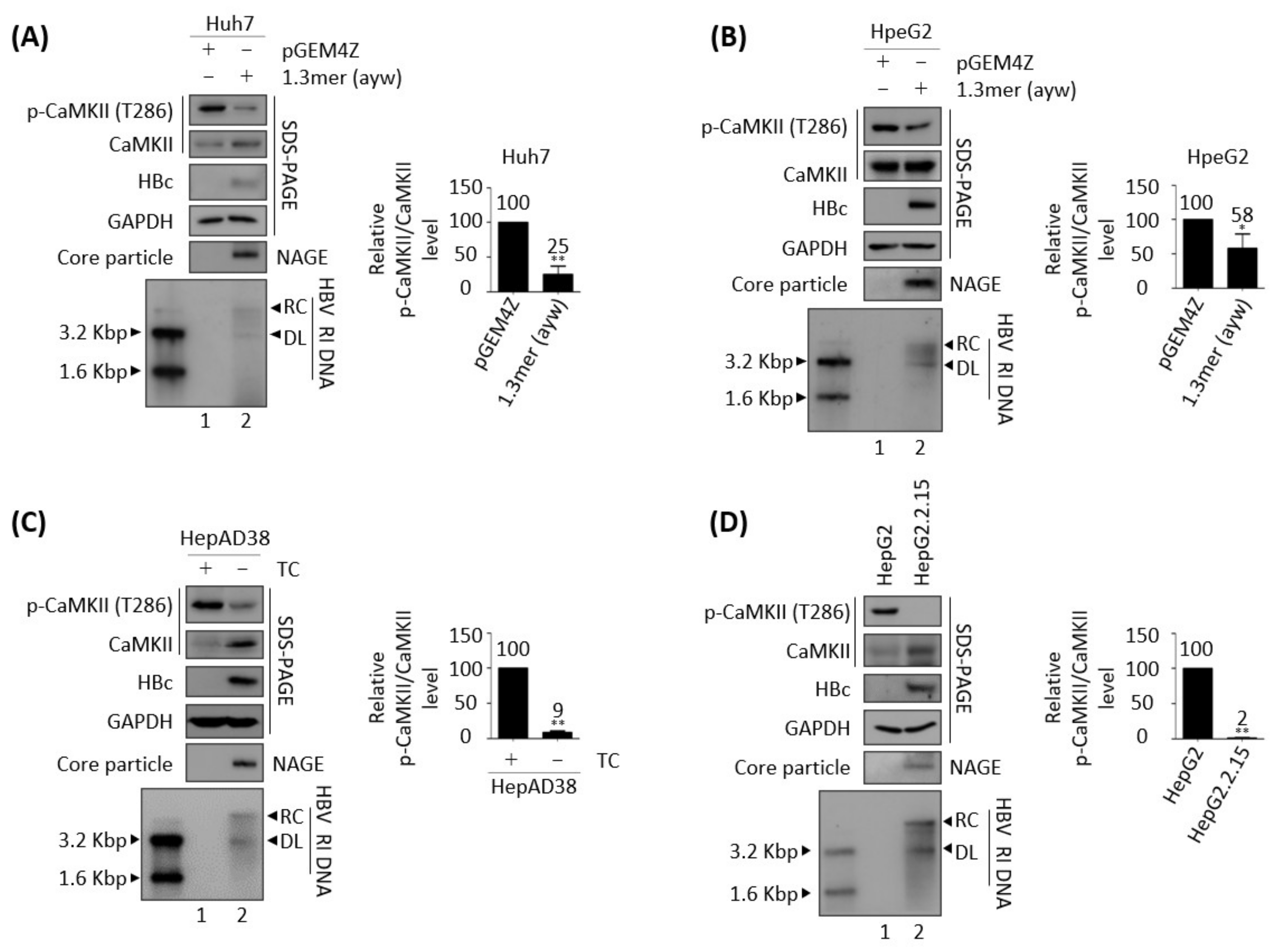
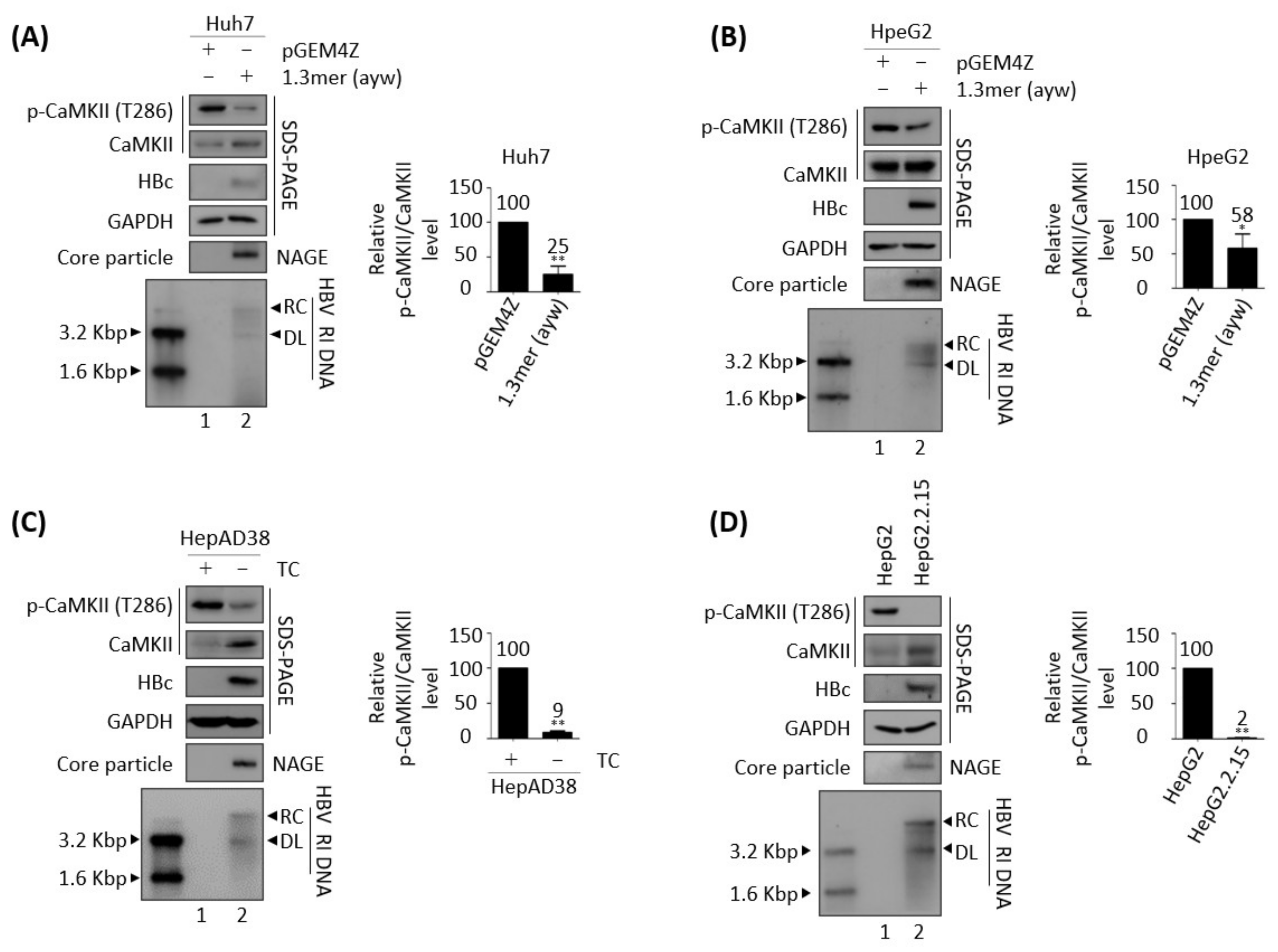
3.1. Active, Phosphorylated CaMKII Is Decreased in HBV Replicating Cells
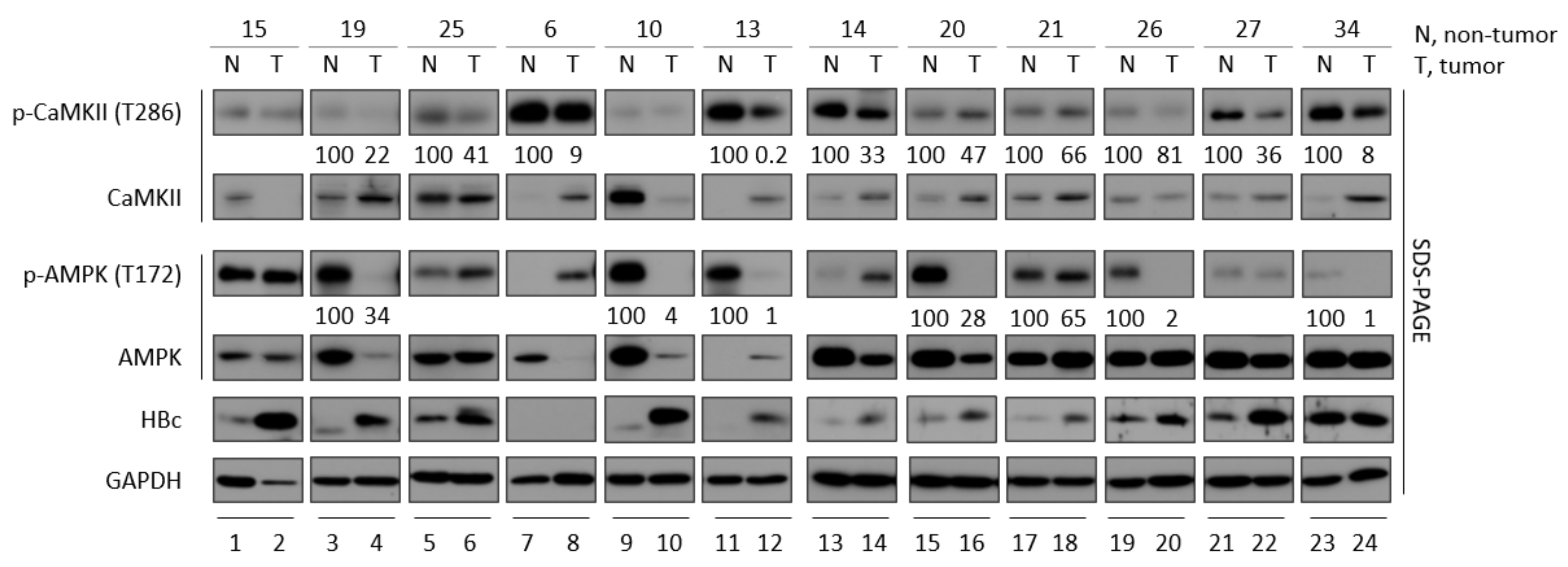
3.2. CaMKII and AMPK Activities Tend to Be Lower in Tumor Tissues than Adjacent Non-Tumor Tissues from Patients with HBV-Associated HCC
3.3. Effects of HBV Replication on CaMKII, AMPK, and AKT/mTOR Phosphorylation
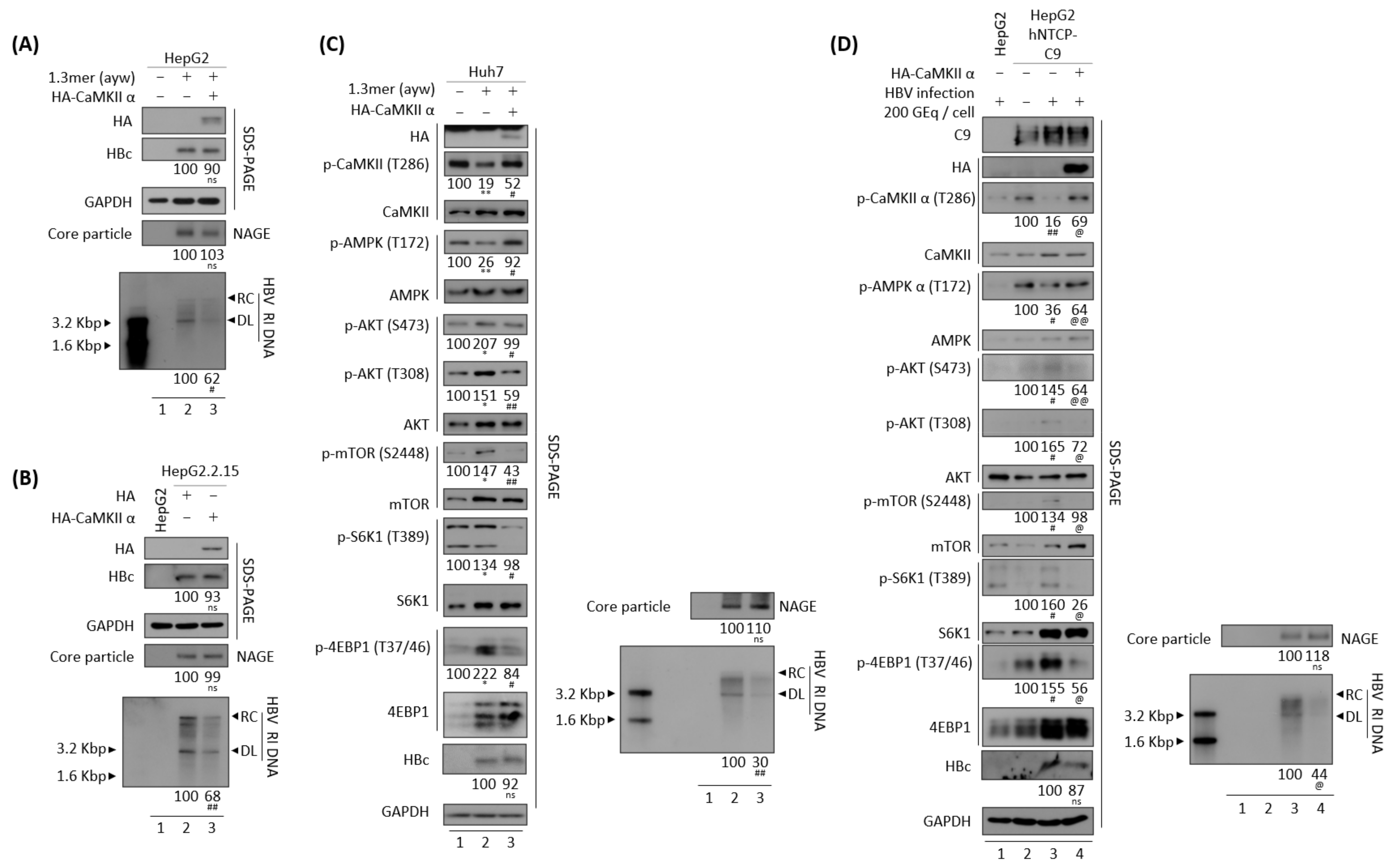
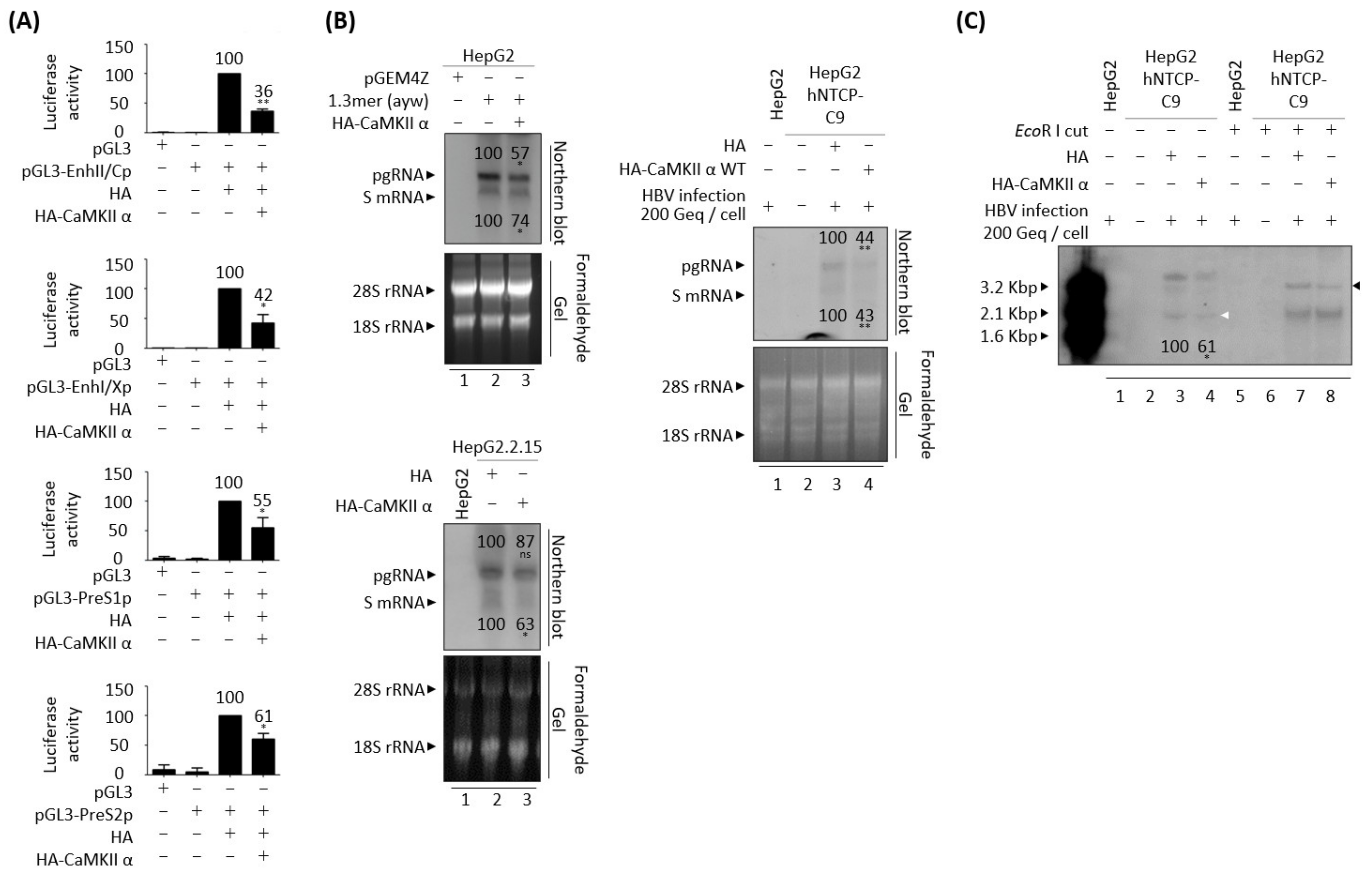
3.4. CaMKII Overexpression Inhibits the HBV Replication through AKT/mTOR Signaling
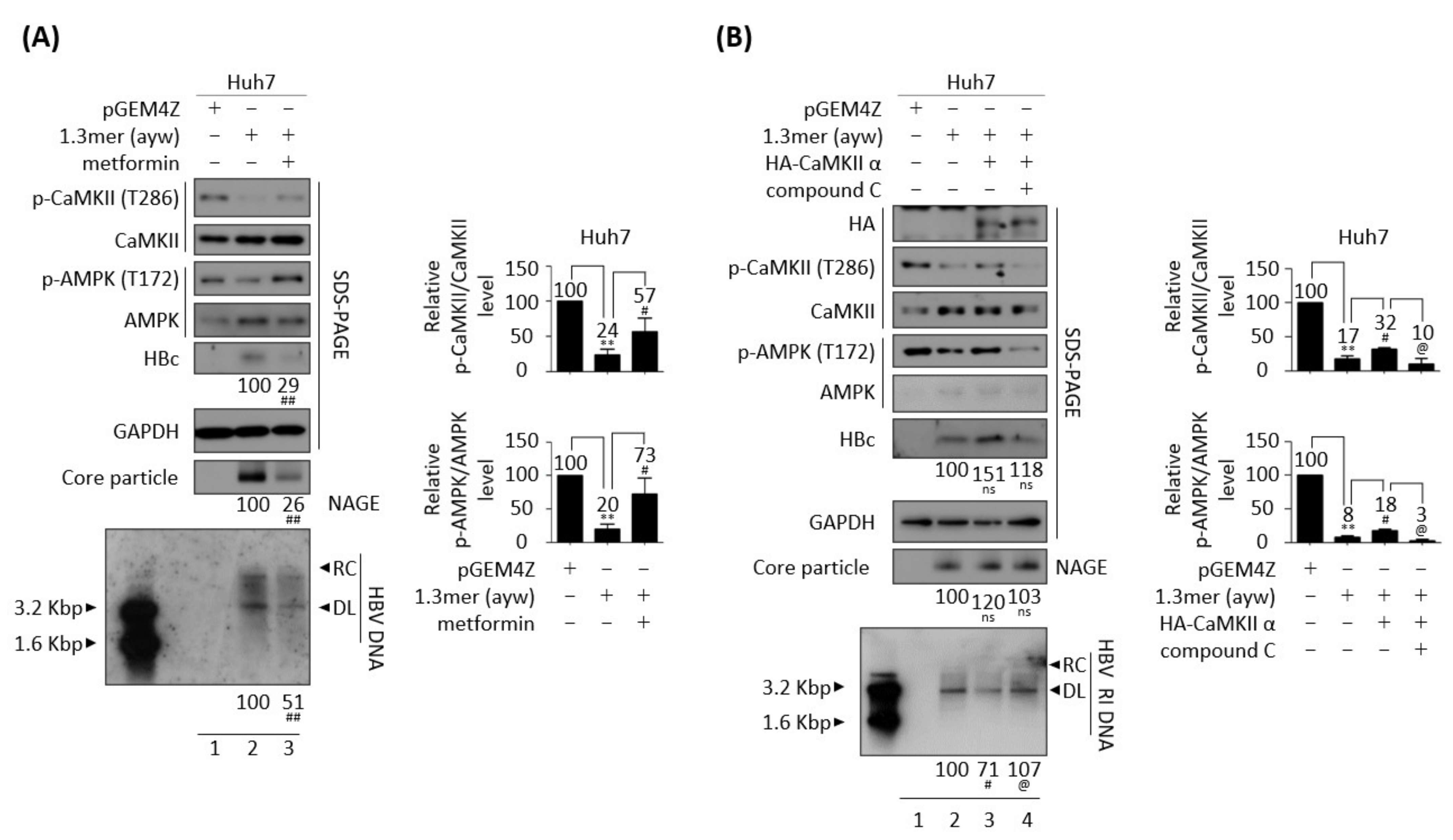
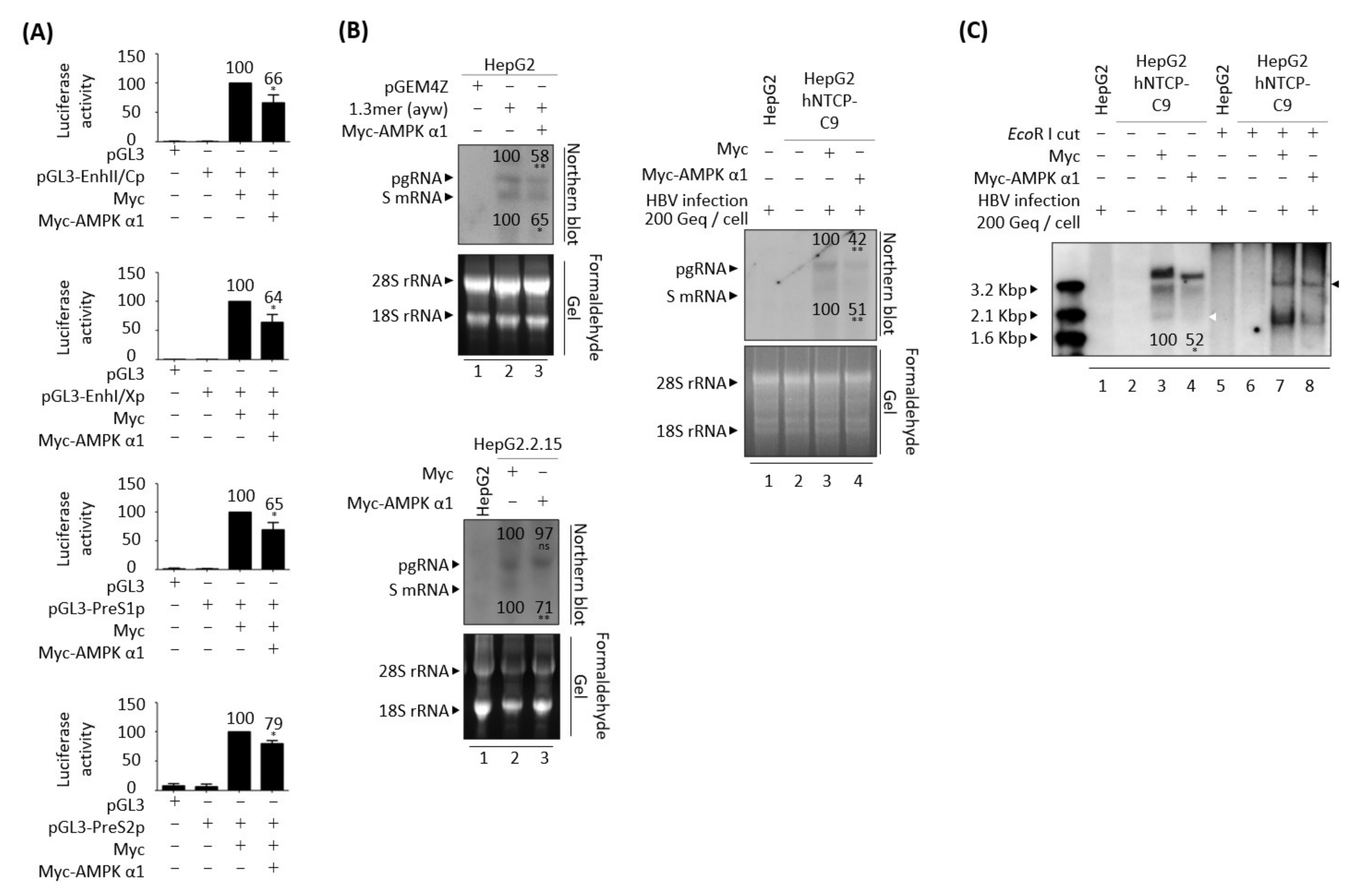
3.5. AMPK Overexpression or Activation Inhibits HBV Replication
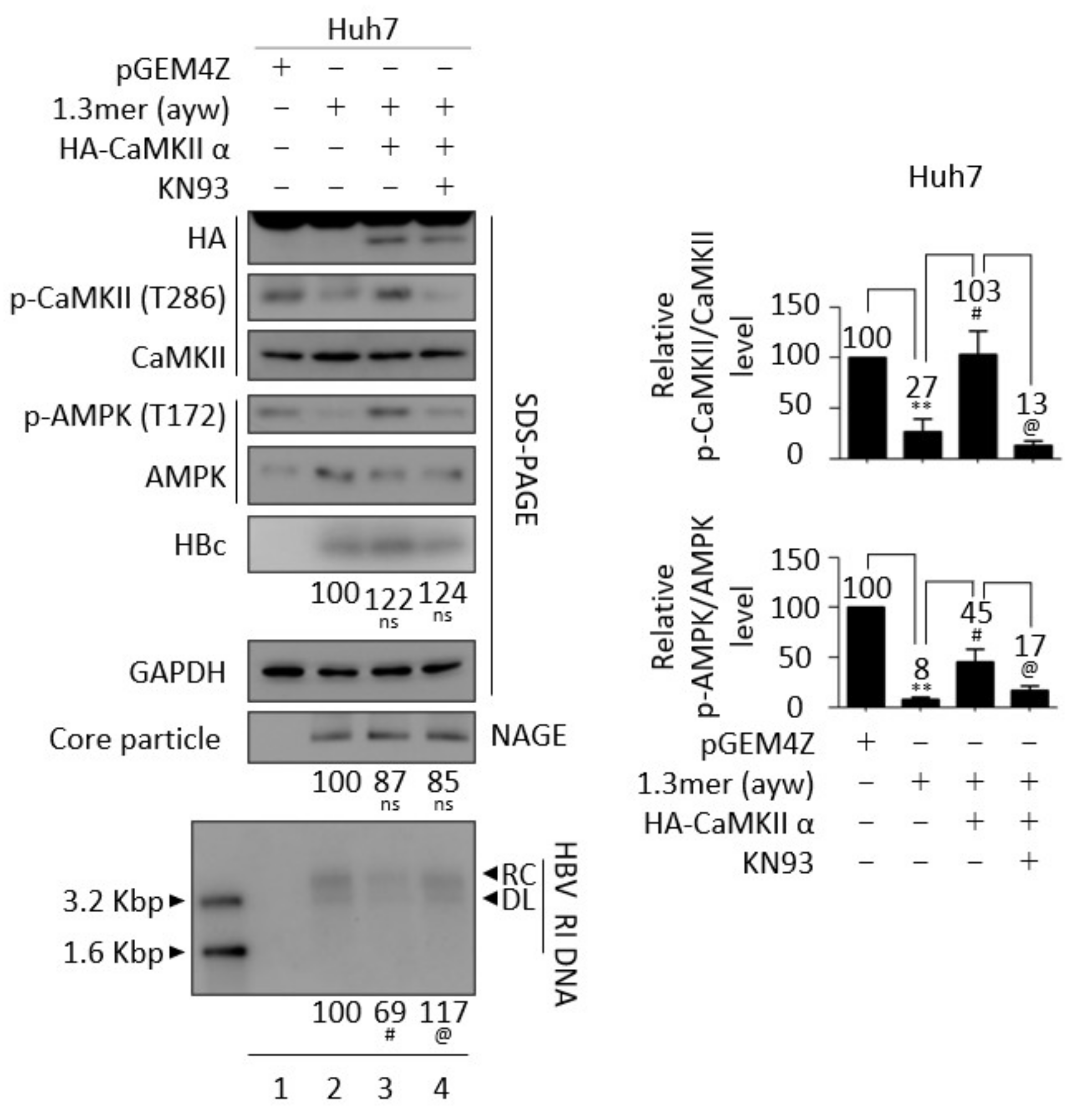
3.6. Inhibition of CaMKII Enhances HBV Replication
3.7. CaMKII and AMPK Form a Feedback Loop
3.8. CaMKII Overexpression Reduces HBV RNAs Due to the Decreased HBV cccDNA
3.9. AMPK Overexpression Reduces HBV RNAs Due to the Decreased HBV cccDNA
3.10. Reduced HBV Replication by Overexpression of CaMKII or AMPK Is HBx Independent
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Piracha, Z.Z.; Saeed, U.; Kim, J.; Kwon, H.; Chwae, Y.J.; Lee, H.W.; Lim, J.H.; Park, S.; Shin, H.J.; Kim, K. An Alternatively Spliced Sirtuin 2 Isoform 5 Inhibits Hepatitis B Virus Replication from cccDNA by Repressing Epigenetic Modifications Made by Histone Lysine Methyltransferases. J. Virol. 2020, 94, e00926-20. [Google Scholar] [CrossRef]
- Cattaneo, R.; Will, H.; Schaller, H. Hepatitis B virus transcription in the infected liver. EMBO J. 1984, 3, 2191–2196. [Google Scholar] [CrossRef]
- Su, T.S.; Lui, W.Y.; Lin, L.H.; Han, S.H.; P’Eng, F.K. Analysis of hepatitis B virus transcripts in infected human livers. Hepatology 1989, 9, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.J.; Li, H.Y.; Chen, Z.H.; Zhou, W.J.; Li, J.J.; Zhang, J.Y.; Wang, J.; Luo, X.Y.; Zeng, T.; Shi, Z.; et al. NAMPT promotes hepatitis B virus replication and liver cancer cell proliferation through the regulation of aerobic glycolysis. Oncol. Lett. 2021, 21, 390. [Google Scholar] [CrossRef]
- Wu, Y.H.; Yang, Y.; Chen, C.H.; Hsiao, C.J.; Li, T.N.; Liao, K.J.; Watashi, K.; Chen, B.S.; Wang, L.H. Aerobic glycolysis supports hepatitis B virus protein synthesis through interaction between viral surface antigen and pyruvate kinase isoform M2. PLoS Pathog. 2021, 17, e1008866. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, R.; Fang, P.; Li, M.; Yu, H.; Wang, Q.; Yu, Y.; Wang, F.; Zhang, Y.; Chen, A.; et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat. Commun. 2021, 12, 98. [Google Scholar] [CrossRef]
- Sangineto, M.; Villani, R.; Cavallone, F.; Romano, A.; Loizzi, D.; Serviddio, G. Lipid Metabolism in Development and Progression of Hepatocellular Carcinoma. Cancers 2020, 12, 1419. [Google Scholar] [CrossRef]
- Hu, B.; Lin, J.Z.; Yang, X.B.; Sang, X.T. Aberrant lipid metabolism in hepatocellular carcinoma cells as well as immune microenvironment: A review. Cell Prolif. 2020, 53, e12772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates with the Risk of Developing Liver Cancer. Front. Cell Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xun, Y.H.; Zhang, Y.J.; Pan, Q.C.; Mao, R.C.; Qin, Y.L.; Liu, H.Y.; Zhang, Y.M.; Yu, Y.S.; Tang, Z.H.; Lu, M.J.; et al. Metformin inhibits hepatitis B virus protein production and replication in human hepatoma cells. J. Viral Hepat. 2014, 21, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Yuan, K.; Zhou, L.; Wang, K.; Chen, H.N.; Lei, Y.; Lan, J.; Pu, Q.; Gao, W.; Zhang, L.; et al. PRKAA/AMPK restricts HBV replication through promotion of autophagic degradation. Autophagy 2016, 12, 1507–1520. [Google Scholar] [CrossRef] [Green Version]
- Xiang, K.; Wang, B. Role of the PI3K-AKT-mTOR pathway in hepatitis B virus infection and replication. Mol. Med. Rep. 2018, 17, 4713–4719. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y.; Kemper, T.; Chen, J.; Yuan, Z.; Liu, S.; Zhu, Y.; Broering, R.; Lu, M. AMPK and Akt/mTOR signalling pathways participate in glucose-mediated regulation of hepatitis B virus replication and cellular autophagy. Cell. Microbiol. 2020, 22, e13131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.G.; Kennedy, M.B. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: A Ca2+-triggered molecular switch. Cell 1986, 44, 861–870. [Google Scholar] [CrossRef]
- Wang, S.Q.; Liu, J.; Qin, J.; Zhu, Y.; Tin, V.P.; Yam, J.W.P.; Wong, M.P.; Xiao, Z.J. CAMK2A supported tumor initiating cells of lung adenocarcinoma by upregulating SOX2 through EZH2 phosphorylation. Cell Death Dis. 2020, 11, 410. [Google Scholar] [CrossRef]
- Bayer, K.U.; Löhler, J.; Schulman, H.; Harbers, K. Developmental expression of the CaM kinase II isoforms: Ubiquitous gamma- and delta-CaM kinase II are the early isoforms and most abundant in the developing nervous system. Mol. Brain Res. 1999, 70, 147–154. [Google Scholar] [CrossRef]
- Tobimatsu, T.; Fujisawa, H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J. Biol. Chem. 1989, 264, 17907–17912. [Google Scholar] [CrossRef]
- Bennett, M.K.; Kennedy, M.B. Deduced primary structure of the beta subunit of brain type II Ca2+/calmodulin-dependent protein kinase determined by molecular cloning. Proc. Natl. Acad. Sci. USA 1987, 84, 1794–1798. [Google Scholar] [CrossRef] [Green Version]
- Hudmon, A.; Schulman, H. Neuronal CA2+/calmodulin-dependent protein kinase II: The role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 2002, 71, 473–510. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Nairn, A.C.; Kuriyan, J. Structural basis for the autoinhibition of calcium/calmodulin-dependent protein kinase I. Cell 1996, 84, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Kanaseki, T.; Ikeuchi, Y.; Sugiura, H.; Yamauchi, T. Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy. J. Cell Biol. 1991, 115, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.B.; Zaegel, V.; Coultrap, S.J.; Miller, A.P.; Bayer, K.U.; Reichow, S.L. The CaMKII holoenzyme structure in activation-competent conformations. Nat. Commun. 2017, 8, 15742. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Orfila, J.E.; Dietz, R.M.; Moreno-Garcia, M.; Rodgers, K.M.; Coultrap, S.J.; Quillinan, N.; Traystman, R.J.; Bayer, K.U.; Herson, P.S. Autonomous CaMKII Activity as a Drug Target for Histological and Functional Neuroprotection after Resuscitation from Cardiac Arrest. Cell Rep. 2017, 18, 1109–1117. [Google Scholar] [CrossRef]
- Cook, S.G.; Buonarati, O.R.; Coultrap, S.J.; Bayer, K.U. CaMKII holoenzyme mechanisms that govern the LTP versus LTD decision. Sci. Adv. 2021, 7, eabe2300. [Google Scholar] [CrossRef]
- Shi, C.; Cai, Y.; Li, Y.; Li, Y.; Hu, N.; Ma, S.; Hu, S.; Zhu, P.; Wang, W.; Zhou, H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018, 14, 59–71. [Google Scholar] [CrossRef]
- Huang, T.; Xu, S.; Deo, R.; Ma, A.; Li, H.; Ma, K.; Gan, X. Targeting the Ca(2+)/Calmodulin-dependent protein kinase II by Tetrandrine in human liver cancer cells. Biochem. Biophys. Res. Commun. 2019, 508, 1227–1232. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Piracha, Z.Z.; Kwon, H.; Saeed, U.; Kim, J.; Jung, J.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Sirtuin 2 Isoform 1 Enhances Hepatitis B Virus RNA Transcription and DNA Synthesis through the AKT/GSK-3β/β-Catenin Signaling Pathway. J. Virol. 2018, 92, e00955-18. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Lee, J.; Lee, S.; Kwak, D.; Choe, W.; Kang, I.; Kim, S.S.; Ha, J. Krill Oil Supplementation Improves Dyslipidemia and Lowers Body Weight in Mice Fed a High-Fat Diet Through Activation of AMP-Activated Protein Kinase. J. Med. Food 2016, 19, 1120–1129. [Google Scholar] [CrossRef]
- Saeed, U.; Kim, J.; Piracha, Z.Z.; Kwon, H.; Jung, J.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Parvulin 14 and Parvulin 17 Bind to HBx and cccDNA and Upregulate Hepatitis B Virus Replication from cccDNA to Virion in an HBx-Dependent Manner. J. Virol. 2019, 93, e01840-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.C.; Chen, Y.H.; Kao, J.H.; Ching, C.; Liu, I.J.; Wang, C.C.; Tsai, C.H.; Wu, F.Y.; Liu, C.J.; Chen, P.J.; et al. Permanent Inactivation of HBV Genomes by CRISPR/Cas9-Mediated Non-cleavage Base Editing. Mol. Ther. Nucleic Acids 2020, 20, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 1005–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severi, T.; Ying, C.; Vermeesch, J.R.; Cassiman, D.; Cnops, L.; Verslype, C.; Fevery, J.; Arckens, L.; Neyts, J.; van Pelt, J.F. Hepatitis B virus replication causes oxidative stress in HepAD38 liver cells. Mol. Cell. Biochem. 2006, 290, 79–85. [Google Scholar] [CrossRef]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nkongolo, S.; Ni, Y.; Lempp, F.A.; Kaufman, C.; Lindner, T.; Esser-Nobis, K.; Lohmann, V.; Mier, W.; Mehrle, S.; Urban, S. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J. Hepatol. 2014, 60, 723–731. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Jung, J.; Kim, H.Y.; Kim, T.; Shin, B.H.; Park, G.S.; Park, S.; Chwae, Y.J.; Shin, H.J.; Kim, K. C-terminal substitution of HBV core proteins with those from DHBV reveals that arginine-rich 167RRRSQSPRR175 domain is critical for HBV replication. PLoS ONE 2012, 7, e41087. [Google Scholar] [CrossRef]
- Kim, H.Y.; Park, G.S.; Kim, E.G.; Kang, S.H.; Shin, H.J.; Park, S.; Kim, K.H. Oligomer synthesis by priming deficient polymerase in hepatitis B virus core particle. Virology 2004, 322, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.; Lee, S.; Windisch, M.P.; Ryu, W.S. DDX3 DEAD-box RNA helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J. Virol. 2014, 88, 13689–13698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Urban, S. Hepatitis B Virus Infection of HepaRG Cells, HepaRG-hNTCP Cells, and Primary Human Hepatocytes. Methods Mol. Biol. 2017, 1540, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef]
- Cai, D.; Nie, H.; Yan, R.; Guo, J.T.; Block, T.M.; Guo, H. A southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol. Biol. 2013, 1030, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Kim, N.K.; Park, S.; Shin, H.J.; Hwang, S.G.; Kim, K. Inhibitory effect of Phyllanthus urinaria L. extract on the replication of lamivudine-resistant hepatitis B virus In Vitro. BMC Complement. Altern. Med. 2015, 15, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meineke, R.; Rimmelzwaan, G.F.; Elbahesh, H. Influenza Virus Infections and Cellular Kinases. Viruses 2019, 11, 171. [Google Scholar] [CrossRef] [Green Version]
- Mesubi, O.O.; Anderson, M.E. Atrial remodelling in atrial fibrillation: CaMKII as a nodal proarrhythmic signal. Cardiovasc. Res. 2016, 109, 542–557. [Google Scholar] [CrossRef] [Green Version]
- Raney, M.A.; Turcotte, L.P. Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. J. Appl. Physiol. 2008, 104, 1366–1373. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.J.; Lin, Y.J.; Yen, C.S.; Tsai, H.W.; Tsai, T.F.; Chang, K.Y.; Huang, W.C.; Lin, P.W.; Chiang, C.W.; Chang, T.T. Hepatitis B virus X protein upregulates mTOR signaling through IKKβ to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS ONE 2012, 7, e41931. [Google Scholar] [CrossRef]
- Kim, A.S.; Miller, E.J.; Young, L.H. AMP-activated protein kinase: A core signalling pathway in the heart. Acta Physiol. 2009, 196, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzatsos, A.; Tsichlis, P.N. Energy depletion inhibits phosphatidylinositol 3-kinase/Akt signaling and induces apoptosis via AMP-activated protein kinase-dependent phosphorylation of IRS-1 at Ser-794. J. Biol. Chem. 2007, 282, 18069–18082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melegari, M.; Scaglioni, P.P.; Wands, J.R. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J. Virol. 1998, 72, 1737–1743. [Google Scholar] [CrossRef] [Green Version]
- You, X.; Liu, F.; Zhang, T.; Li, Y.; Ye, L.; Zhang, X. Hepatitis B virus X protein upregulates oncogene Rab18 to result in the dysregulation of lipogenesis and proliferation of hepatoma cells. Carcinogenesis 2013, 34, 1644–1652. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Shin, H.J.; Kim, K.; Choi, H.M.; Rhee, S.H.; Moon, H.B.; Kim, H.H.; Yang, U.S.; Yu, D.Y.; Cheong, J. Hepatitis B virus X protein induces hepatic steatosis via transcriptional activation of SREBP1 and PPARgamma. Gastroenterology 2007, 132, 1955–1967. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Rawat, S.; Ajenjo, M.; Bouchard, M.J. Hepatitis B virus (HBV) X protein-mediated regulation of hepatocyte metabolic pathways affects viral replication. Virology 2016, 498, 9–22. [Google Scholar] [CrossRef]
- Wang, M.D.; Wu, H.; Huang, S.; Zhang, H.L.; Qin, C.J.; Zhao, L.H.; Fu, G.B.; Zhou, X.; Wang, X.M.; Tang, L.; et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget 2016, 7, 6711–6726. [Google Scholar] [CrossRef]
- Jiang, X.; Tan, H.Y.; Teng, S.; Chan, Y.T.; Wang, D.; Wang, N. The Role of AMP-Activated Protein Kinase as a Potential Target of Treatment of Hepatocellular Carcinoma. Cancers 2019, 11, 647. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; Larocca, M.C.; Favre, C. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef]
- Luo, Y.D.; Fang, L.; Yu, H.Q.; Zhang, J.; Lin, X.T.; Liu, X.Y.; Wu, D.; Li, G.X.; Huang, D.; Zhang, Y.J.; et al. p53 haploinsufficiency and increased mTOR signalling define a subset of aggressive hepatocellular carcinoma. J. Hepatol. 2021, 74, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, X.F.; Wang, N.Y.; Wang, X.M.; Liang, S.T.; Zheng, W.; Lu, Y.B.; Zhao, X.; Hao, D.L.; Zhang, Z.Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, G.; Davaakhuu, G.; Kaplun, L.; Chung, W.C.; Rana, A.; Atfi, A.; Miele, L.; Tzivion, G. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J. Biol. Chem. 2014, 289, 6054–6066. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Hwang, S.G.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J. Virol. 2014, 88, 8754–8767. [Google Scholar] [CrossRef] [Green Version]
- Basagoudanavar, S.H.; Perlman, D.H.; Hu, J. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J. Virol. 2007, 81, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- De Rocquigny, H.; Rat, V.; Pastor, F.; Darlix, J.L.; Hourioux, C.; Roingeard, P. Phosphorylation of the Arginine-Rich C-Terminal Domains of the Hepatitis B Virus (HBV) Core Protein as a Fine Regulator of the Interaction between HBc and Nucleic Acid. Viruses 2020, 12, 738. [Google Scholar] [CrossRef]
- Selzer, L.; Kant, R.; Wang, J.C.; Bothner, B.; Zlotnick, A. Hepatitis B Virus Core Protein Phosphorylation Sites Affect Capsid Stability and Transient Exposure of the C-terminal Domain. J. Biol. Chem. 2015, 290, 28584–28593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Böttcher, B.; Nassal, M. Hepatitis B virus core protein phosphorylation: Identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog. 2018, 14, e1007488. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.; Basagoudanavar, S.H.; Liu, K.; Luckenbaugh, L.; Wei, D.; Wang, C.; Wei, B.; Zhao, Y.; Yan, T.; Delaney, W.; et al. Capsid Phosphorylation State and Hepadnavirus Virion Secretion. J. Virol. 2017, 91, e00092-17. [Google Scholar] [CrossRef] [Green Version]
- White, R.R.; Kwon, Y.G.; Taing, M.; Lawrence, D.S.; Edelman, A.M. Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J. Biol. Chem. 1998, 273, 3166–3172. [Google Scholar] [CrossRef] [Green Version]
- Kennelly, P.J.; Krebs, E.G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 1991, 266, 15555–15558. [Google Scholar] [CrossRef]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Edman, C.F.; Schulman, H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J. Cell Biol. 1994, 126, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Brocke, L.; Srinivasan, M.; Schulman, H. Developmental and regional expression of multifunctional Ca2+/calmodulin-dependent protein kinase isoforms in rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 6797–6808. [Google Scholar] [CrossRef] [Green Version]
- Diogo Dias, J.; Sarica, N.; Neuveut, C. Early Steps of Hepatitis B Life Cycle: From Capsid Nuclear Import to cccDNA Formation. Viruses 2021, 13, 757. [Google Scholar] [CrossRef]
- Yu, D.Y. Relevance of reactive oxygen species in liver disease observed in transgenic mice expressing the hepatitis B virus X protein. Lab. Anim. Res. 2020, 36, 6. [Google Scholar] [CrossRef] [PubMed]
- Kodiha, M.; Rassi, J.G.; Brown, C.M.; Stochaj, U. Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK-->ERK1/2 pathway. Am. J. Physiol. Cell Physiol. 2007, 293, C1427–C1436. [Google Scholar] [CrossRef]
- Vats, R.; Li, Z.; Ju, E.M.; Dubey, R.K.; Kaminski, T.W.; Watkins, S.; Pradhan-Sundd, T. Intravital imaging reveals inflammation as a dominant pathophysiology of age-related hepatovascular changes. Am. J. Physiol. Cell Physiol. 2022, in press. [CrossRef]
- Kemp, L.; Clare, K.E.; Brennan, P.N.; Dillon, J.F. New horizons in hepatitis B and C in the older adult. Age Ageing 2019, 48, 32–37. [Google Scholar] [CrossRef]
- Vieira, R.F.L.; Junqueira, R.L.; Gaspar, R.C.; Muñoz, V.R.; Pauli, J.R. Exercise activates AMPK signaling: Impact on glucose uptake in the skeletal muscle in aging. J. Rehabil. Ther. 2020, 2, 48–53. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Kwon, H.; Kalsoom, F.; Sajjad, M.A.; Lee, H.W.; Lim, J.H.; Jung, J.; Chwae, Y.-J.; Park, S.; Shin, H.-J.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II Inhibits Hepatitis B Virus Replication from cccDNA via AMPK Activation and AKT/mTOR Suppression. Microorganisms 2022, 10, 498. https://doi.org/10.3390/microorganisms10030498
Kim J, Kwon H, Kalsoom F, Sajjad MA, Lee HW, Lim JH, Jung J, Chwae Y-J, Park S, Shin H-J, et al. Ca2+/Calmodulin-Dependent Protein Kinase II Inhibits Hepatitis B Virus Replication from cccDNA via AMPK Activation and AKT/mTOR Suppression. Microorganisms. 2022; 10(3):498. https://doi.org/10.3390/microorganisms10030498
Chicago/Turabian StyleKim, Jumi, Hyeonjoong Kwon, Fadia Kalsoom, Muhammad Azhar Sajjad, Hyun Woong Lee, Jin Hong Lim, Jaesung Jung, Yong-Joon Chwae, Sun Park, Ho-Joon Shin, and et al. 2022. "Ca2+/Calmodulin-Dependent Protein Kinase II Inhibits Hepatitis B Virus Replication from cccDNA via AMPK Activation and AKT/mTOR Suppression" Microorganisms 10, no. 3: 498. https://doi.org/10.3390/microorganisms10030498