
Resistance Is Not Futile: The Role of Quorum Sensing Plasticity in Pseudomonas aeruginosa Infections and Its Link to Intrinsic Mechanisms of Antibiotic Resistance


Abstract
:1. Introduction
2. Clinical Ubiquity and Transmission
2.1. P. aeruginosa and Pulmonary Infections
2.2. P. aeruginosa Is More Than Just a Lung Pathogen
2.2.1. Burn Wounds
2.2.2. UTIs
3. Pathogenicity and Virulence
3.1. Quorum Sensing (QS): For the Many, Not the Few
3.2. The Complex Regulatory Network of QS in P. aeruginosa: The Key Determinants to QS Progression
3.3. The Rhl System Can Bypass the Requirement for Activation by the Las System
- P. aeruginosa is found in several different environments, both inside and outside the host. Due to its large genome, it is highly adaptable to thrive under different conditions and stresses. Is there a standard growth condition for laboratory experiments that represents a viable way to assess virulence factor production that is more clinically relevant?
- A variety of host infection models exist for most of the clinically relevant infections caused by P. aeruginosa. How closely do these resemble the human host? What are the host factors that complicate understanding P. aeruginosa pathogenesis?
- The efficacy of anti-QS therapies has been assessed in vitro with modest effects but has failed at limiting virulence in vivo. What are the gaps in our understanding of the system that result in this disconnect? Have our laboratory strains been tailored to meet our scientific research questions but are no longer capable of addressing our clinical needs?
3.4. QS-Driven Pathogenesis Proceeds through RhlR and PqsE in a PQS-Independent Manner
3.5. Disease Relevant Virulence Factors
3.5.1. Elastase
3.5.2. Pyocyanin
3.5.3. Alginate, Psl, and Pel
3.5.4. Hydrogen Cyanide
3.6. Microevolution of P. aeruginosa in Pulmonary Infections
3.7. The Role of QS in UTIs and Bound Wounds
4. Intrinsic Antibiotic Resistance Mechanisms
4.1. Biofilms
4.2. Outer Membrane Porins
4.3. Efflux Pumps
5. Future Prospects & Conclusions
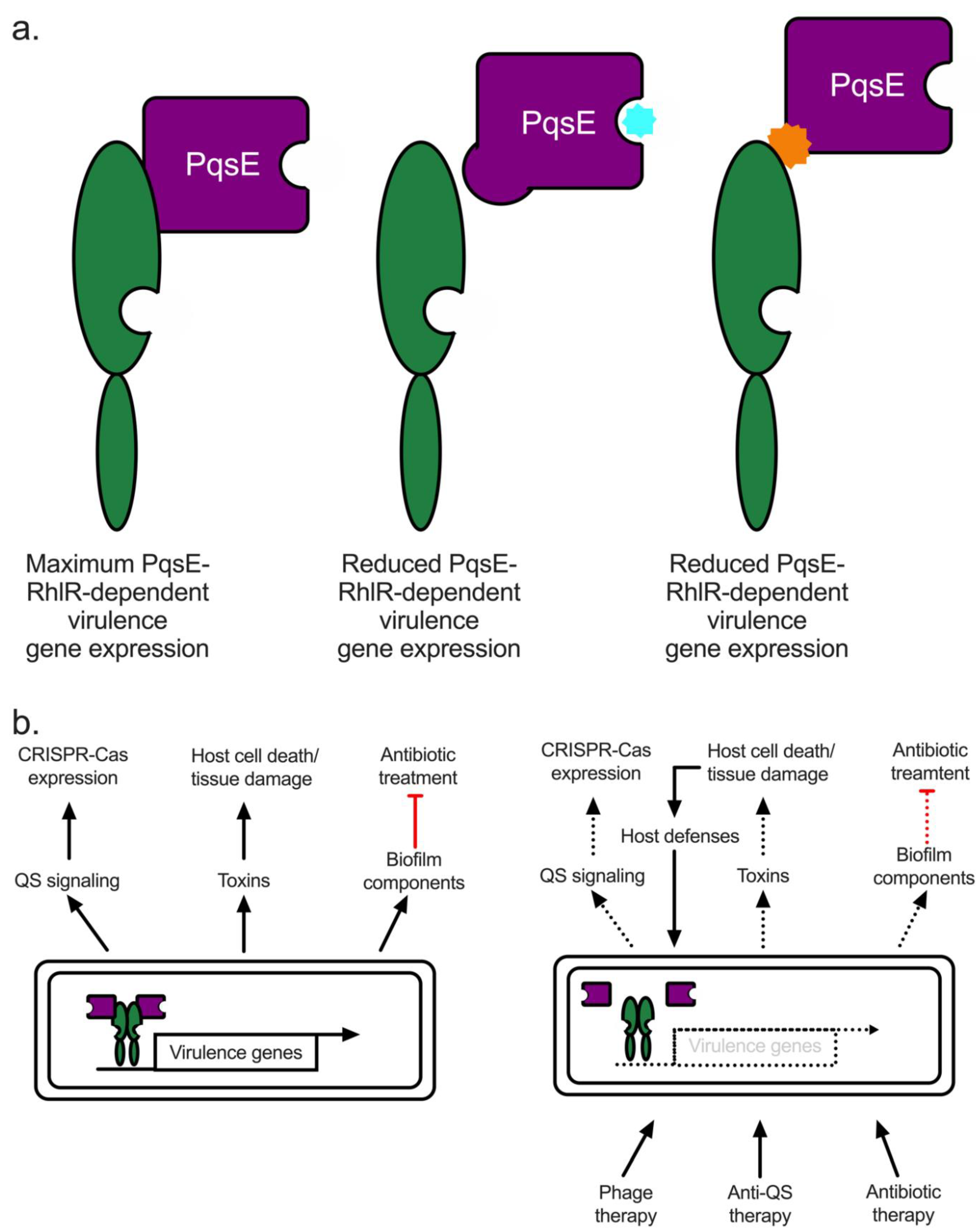
5.1. Quorum Quenching in P. aeruginosa: The PqsE-RhlR Interaction Represents Renewed Hope for a Viable Anti-QS Therapeutic
5.2. Alternative Therapeutics: Phage and Antibody Therapies
5.3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Redfield, R.R. Antibiotic Resistance Threats in the United States; Centres for Disease Control and Prevention: Atlanta, GA, USA, 2019. [Google Scholar]
- Centers for Disease Control and Prevention. Biggest Threats and Data: 2019 AR Threats Report; CDC: Atlanta, GA, USA, 2019. [Google Scholar]
- Bjarnsholt, T.; Jensen, P.O.; Jakobsen, T.H.; Phipps, R.; Nielsen, A.K.; Rybtke, M.T.; Tolker-Nielsen, T.; Givskov, M.; Hoiby, N.; Ciofu, O.; et al. Quorum sensing and virulence of Pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PLoS ONE 2010, 5, e10115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolter, D.J.; Lister, P.D. Mechanisms of beta-lactam resistance among Pseudomonas aeruginosa. Curr. Pharm. Des. 2013, 19, 209–222. Available online: https://www.ncbi.nlm.nih.gov/pubmed/22894618 (accessed on 25 November 2021). [CrossRef] [PubMed]
- Fang, Z.L.; Zhang, L.Y.; Huang, Y.M.; Qing, Y.; Cao, K.Y.; Tian, G.B.; Huang, X. OprD mutations and inactivation in imipenem-resistant Pseudomonas aeruginosa isolates from China. Infect. Genet. Evol. 2014, 21, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Cabot, G.; Zamorano, L.; Moya, B.; Juan, C.; Navas, A.; Blazquez, J.; Oliver, A. Evolution of Pseudomonas aeruginosa Antimicrobial Resistance and Fitness under Low and High Mutation Rates. Antimicrob. Agents Chemother. 2016, 60, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.G.; Parsek, M.R.; Pearson, J.P.; Iglewski, B.H.; Costerton, J.W.; Greenberg, E.P. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 1998, 280, 295–298. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9535661 (accessed on 25 November 2021). [CrossRef] [Green Version]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Drenkard, E.; Ausubel, F.M. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 2002, 416, 740–743. [Google Scholar] [CrossRef]
- Okoliegbe, I.N.; Hijazi, K.; Cooper, K.; Ironside, C.; Gould, I.M. Trends of antimicrobial resistance and combination susceptibility testing of multidrug-resistant Pseudomonas aeruginosa isolates from cystic fibrosis patients: A 10-year update. Antimicrob. Agents Chemother. 2021, 65, e02483-20. [Google Scholar] [CrossRef]
- Hassett, D.J.; Borchers, M.T.; Panos, R.J. Chronic obstructive pulmonary disease (COPD): Evaluation from clinical, immunological and bacterial pathogenesis perspectives. J. Microbiol. 2014, 52, 211–226. [Google Scholar] [CrossRef]
- Montero, M.; Domínguez, M.; Orozco-Levi, M.; Salvadó, M.; Knobel, H. Mortality of COPD patients infected with multi-resistant Pseudomonas aeruginosa: A case and control study. Infection 2009, 37, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorlí, L.; Luque, S.; Gómez-Zorrilla, S.; Benito, N.; Grau, S. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef] [PubMed]
- Kaier, K.; Heister, T.; Götting, T.; Wolkewitz, M.; Mutters, N.T. Measuring the in-hospital costs of Pseudomonas aeruginosa pneumonia: Methodology and results from a German teaching hospital. BMC Infect. Dis. 2019, 19, 1028. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.F.; Brauer, A.L.; Eschberger, K.; Lobbins, P.; Grove, L.; Cai, X.; Sethi, S. Pseudomonas aeruginosa in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 853–860. [Google Scholar] [CrossRef]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 2016, 16, 174. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.K.; Rau, M.H.; Johansen, H.K.; Ciofu, O.; Jelsbak, L.; Yang, L.; Folkesson, A.; Jarmer, H.Ø.; Aanæs, K.; Von Buchwald, C.; et al. Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. ISME J. 2012, 6, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ramezanpour, M.; Fong, S.A.; Cooksley, C.; Murphy, J.; Suzuki, M.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Pseudomonas aeruginosa Exoprotein-Induced Barrier Disruption Correlates With Elastase Activity and Marks Chronic Rhinosinusitis Severity. Front. Cell. Infect. Microbiol. 2019, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Tai, A.S.; Sherrard, L.J.; Kidd, T.J.; Ramsay, K.A.; Buckley, C.; Syrmis, M.; Grimwood, K.; Bell, S.C.; Whiley, D.M. Antibiotic perturbation of mixed-strain Pseudomonas aeruginosa infection in patients with cystic fibrosis. BMC Pulm. Med. 2017, 17, 138. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, P.J.; Izydorcyzk, C.; Clark, S.; Blanchard, A.; Wang, P.W.; Yau, Y.; Waters, V.; Guttman, D.S. Pseudomonas aeruginosa Strain-sharing in Early Infection among Children with Cystic Fibrosis. Clin. Infect. Dis. 2021, 73, E2521–E2528. [Google Scholar] [CrossRef]
- Mayhall, C.G. The epidemiology of burn wound infections: Then and now. Clin. Infect. Dis. 2003, 37, 543–550. [Google Scholar]
- Tredget, E.E.; Shankowsky, H.A.; Rennie, R.; Burrell, R.E.; Logsetty, S. Pseudomonas infections in the thermally injured patient. Burns 2004, 30, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.J.; Hall, C.L.; Schniederberend, M.; Farrow, J.M.; Goodson, J.R.; Pesci, E.C.; Kazmierczak, B.I.; Lee, V.T. Host suppression of quorum sensing during catheter-associated urinary tract infections. Nat. Commun. 2018, 9, 4436. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.W.; Floyd, R.V.; Fothergill, J.L. The contribution of Pseudomonas aeruginosa virulence factors and host factors in the establishment of urinary tract infections. FEMS Microbiol. Lett. 2017, 364, fnx124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, N.; Tempany, E.; Falkiner, F.R.; Fitzgerald, M.X.; O'boyle, C.; Keane, C.T. Does pseudomonas cross-infection occur between cystic-fibrosis patients. Lancet 1982, 320, 688–690. [Google Scholar] [CrossRef]
- Brandenburg, K.S.; Weaver, A.J.; Qian, L.; You, T.; Chen, P.; Karna, S.L.R.; Fourcaudot, A.B.; Sebastian, E.A.; Abercrombie, J.J.; Pineda, U.; et al. Development of Pseudomonas aeruginosa biofilms in partial-thickness burn wounds using a sprague-dawley rat model. J. Burn Care Res. 2019, 40, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, K.S.; Weaver, A.J.; Karna, S.L.R.; You, T.; Chen, P.; Van Stryk, S.; Qian, L.; Pineda, U.; Abercrombie, J.J.; Leung, K.P. Formation of Pseudomonas aeruginosa Biofilms in Full-thickness Scald Burn Wounds in Rats. Sci. Rep. 2019, 9, 13627. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Hauck, C.G.; Weber, D.J.; Cairns, B.A.; Van Duin, D. Bacterial Infections after Burn Injuries: Impact of Multidrug Resistance. Clin. Infect. Dis. 2017, 65, 2130–2136. [Google Scholar] [CrossRef]
- Trafny, E.A. Susceptibility of adherent organisms from Pseudomonas aeruginosa and Staphylococcus aureus strains isolated from burn wounds to antimicrobial agents. Int. J. Antimicrob. Agents 1998, 10, 223–228. [Google Scholar] [CrossRef]
- Paulman, P.M.; Taylor, R.B.; Paulman, A.A.; Nasir, L.S. Family medicine: Principles and Practice; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–1863. [Google Scholar]
- Farhan, N.; Jeffery, S. Diagnosing burn wounds infection: The practice gap & advances with moleculight bacterial imaging. Diagnostics 2021, 11, 268. [Google Scholar]
- Wanis, M.; Walker, S.A.N.; Daneman, N.; Elligsen, M.; Palmay, L.; Simor, A.; Cartotto, R. Impact of hospital length of stay on the distribution of Gram negative bacteria and likelihood of isolating a resistant organism in a Canadian burn center. Burns 2016, 42, 104–111. [Google Scholar] [CrossRef]
- van Duin, D.; Jones, S.W.; Dibiase, L.; Schmits, G.; Lachiewicz, A.; Hultman, C.S.; Rutala, W.A.; Weber, D.J.; Cairns, B.A. Reduction in Central Line–Associated Bloodstream Infections in Patients with Burns. Infect. Control. Hosp. Epidemiol. 2014, 35, 1066–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslova, E.; Shi, Y.; Sjöberg, F.; Azevedo, H.S.; Wareham, D.W.; McCarthy, R.R. An Invertebrate Burn Wound Model That Recapitulates the Hallmarks of Burn Trauma and Infection Seen in Mammalian Models. Front. Microbiol. 2020, 11, 998. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Elgharably, H.; Sinha, M.; Ganesh, K.; Chaney, S.; Mann, E.; Miller, C.; Khanna, S.; Bergdall, V.K.; Powell, H.M.; et al. Mixed-species biofilm compromises wound healing by disrupting epidermal barrier function. J. Pathol. 2014, 233, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.D.; Hultgren, S.J. Urinary tract infections: Microbial pathogenesis, host–pathogen interactions and new treatment strategies. Nat. Rev. Microbiol. 2020, 18, 211–226. [Google Scholar] [CrossRef]
- Mittal, R.; Aggarwal, S.; Sharma, S.; Chhibber, S.; Harjai, K. Urinary tract infections caused by Pseudomonas aeruginosa: A minireview. J. Infect. Public Health 2009, 2, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.J.; Records, A.R.; Orr, M.W.; Linden, S.B.; Lee, V.T. Catheter-associated urinary tract infection by Pseudomonas aeruginosa is mediated by exopolysaccharide-independent biofilms. Infect. Immun. 2014, 82, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Martinez-Yamout, M.A.; Dickerson, T.J.; Brogan, A.P.; Wright, P.E.; Dyson, H.J.; Yamani, L.; Alamri, A.; Alsultan, A.; Alfifi, S.; et al. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. J. Bacteriol. 2014, 9, 2175–2184. [Google Scholar]
- Narten, M.; Rosin, N.; Schobert, M.; Tielen, P. Susceptibility of Pseudomonas aeruginosa urinary tract isolates and influence of urinary tract conditions on antibiotic tolerance. Curr. Microbiol. 2012, 64, 7–16. [Google Scholar] [CrossRef]
- Pearson, J.P.; Passador, L.; Iglewski, B.H.; Greenberg, E.P. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1995, 92, 1490–1494. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7878006 (accessed on 25 November 2021). [CrossRef] [Green Version]
- Winson, M.K.; Camara, M.; Latifi, A.; Foglino, M.; Chhabra, S.R.; Daykin, M.; Bally, M.; Chapon, V.; Salmond, G.P.; Bycroft, B.W.; et al. Multiple N-acyl-L-homoserine lactone signal molecules regulate production of virulence determinants and secondary metabolites in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1995, 92, 9427–9431. Available online: https://www.ncbi.nlm.nih.gov/pubmed/7568146 (accessed on 1 December 2021). [CrossRef] [Green Version]
- Gambello, M.J.; Kaye, S.; Iglewski, B.H. LasR of Pseudomonas aeruginosa is a transcriptional activator of the alkaline protease gene (apr) and an enhancer of exotoxin A expression. Infect. Immun. 1993, 61, 1180–1184. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8454322 (accessed on 1 December 2021). [CrossRef] [PubMed] [Green Version]
- Parsek, M.R.; Val, D.L.; Hanzelka, B.L.; Cronan, J.E.; Greenberg, E.P.; Cronan, J.E., Jr.; Greenberg, E.P. Acyl homoserine-lactone quorum-sensing signal generation. Proc. Natl. Acad. Sci. USA 1999, 96, 4360–4365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brint, J.M.; Ohman, D.E. Synthesis of multiple exoproducts in Pseudomonas aeruginosa is under the control of RhlR-RhlI, another set of regulators in strain PAO1 with homology to the autoinducer-responsive LuxR-LuxI family. J. Bacteriol. 1995, 177, 7155–7163. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8522523 (accessed on 1 December 2021). [CrossRef] [PubMed] [Green Version]
- Pearson, J.P.; Pesci, E.C.; Iglewski, B.H. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J. Bacteriol. 1997, 179, 5756–5767. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9294432 (accessed on 1 December 2021). [CrossRef] [Green Version]
- Brameyer, S.; Heermann, R. Specificity of Signal-Binding via Non-AHL LuxR-Type Receptors. PLoS ONE 2015, 10, e0124093. [Google Scholar] [CrossRef] [Green Version]
- Schuster, M.; Lostroh, C.P.; Ogi, T.; Greenberg, E.P. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: A transcriptome analysis. J. Bacteriol. 2003, 185, 2066–2079. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12644476 (accessed on 1 December 2021). [CrossRef] [Green Version]
- Schuster, M.; Urbanowski, M.L.; Greenberg, E.P. Promoter specificity in Pseudomonas aeruginosa quorum sensing revealed by DNA binding of purified LasR. Proc. Natl. Acad. Sci. USA 2004, 101, 15833–15839. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhang, L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 2014, 6, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Higgins, S.; Heeb, S.; Rampioni, G.; Fletcher, M.P.; Williams, P.; Camara, M. Differential Regulation of the Phenazine Biosynthetic Operons by Quorum Sensing in Pseudomonas aeruginosa PAO1-N. Front. Cell Infect. Microbiol. 2018, 8, 252. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Moustafa, D.; Smith, C.D.; Goldberg, J.B.; Bassler, B.L. The RhlR quorum-sensing receptor controls Pseudomonas aeruginosa pathogenesis and biofilm development independently of its canonical homoserine lactone autoinducer. PLoS Pathog. 2017, 13, e1006504. [Google Scholar] [CrossRef] [Green Version]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, R.; Peschek, N.; Frohlich, K.S.; Schumacher, K.; Papenfort, K. Three autoinducer molecules act in concert to control virulence gene expression in Vibrio cholerae. Nucleic Acids. Res. 2019, 47, 3171–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, W.L.; Bassler, B.L. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacikova, G.; Lin, W.; Skorupski, K. The virulence activator AphA links quorum sensing to pathogenesis and physiology in Vibrio cholerae by repressing the expression of a penicillin amidase gene on the small chromosome. J. Bacteriol. 2003, 185, 4825–4836. Available online: https://www.ncbi.nlm.nih.gov/pubmed/12897002 (accessed on 5 January 2022). [CrossRef] [Green Version]
- Shao, Y.; Bassler, B.L. Quorum regulatory small RNAs repress type VI secretion in Vibrio cholerae. Mol. Microbiol. 2014, 92, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Kovacikova, G.; Skorupski, K. The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol. Microbiol. 2007, 64, 953–967. [Google Scholar] [CrossRef]
- Shao, Y.; Bassler, B.L. Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol. Microbiol. 2012, 83, 599–611. [Google Scholar] [CrossRef]
- Freeman, J.A.; Bassler, B.L. Sequence and function of LuxU: A two-component phosphorelay protein that regulates quorum sensing in Vibrio harveyi. J. Bacteriol. 1999, 181, 899–906. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9922254 (accessed on 5 January 2022). [CrossRef] [Green Version]
- Zhu, J.; Miller, M.B.; Vance, R.E.; Dziejman, M.; Bassler, B.L.; Mekalanos, J.J. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 2002, 99, 3129–3134. [Google Scholar] [CrossRef] [Green Version]
- McGrath, S.; Wade, D.S.; Pesci, E.C. Dueling quorum sensing systems in Pseudomonas aeruginosa control the production of the Pseudomonas quinolone signal (PQS). FEMS Microbiol. Lett. 2004, 230, 27–34. [Google Scholar] [CrossRef]
- Diggle, S.P.; Winzer, K.; Chhabra, S.R.; Worrall, K.E.; Cámara, M.; Williams, P. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 2003, 50, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.S.; Calfee, M.W.; Rocha, E.R.; Ling, E.A.; Engstrom, E.; Coleman, J.P.; Pesci, E.C. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 4372–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, G.; He, J.; Rahme, L.G. Mutation analysis of the Pseudomonas aeruginosa mvfR and pqsABCDE gene promoters demonstrates complex quorum-sensing circuitry. Microbiology 2006, 152, 1679–1686. [Google Scholar] [CrossRef] [Green Version]
- McCready, A.R.; Paczkowski, J.E.; Cong, J.-P.; Bassler, B.L. An autoinducer-independent rhlr quorum-sensing receptor enables analysis of rhlr regulation. PLoS Pathog. 2019, 15, e1007820. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, S.; Pustelny, C.; Ritter, C.; Klinkert, B.; Narberhaus, F.; Häussler, S. The PqsR and RhlR transcriptional regulators determine the level of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa by producing two different pqsABCDE mRNA Isoforms. J. Bacteriol. 2014, 196, 4163–4171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, M.; Hawkins, A.C.; Harwood, C.S.; Greenberg, E.P. The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol. Microbiol. 2004, 51, 973–985. [Google Scholar] [CrossRef]
- Whiteley, M.; Parsek, M.R.; Greenberg, E.P. Regulation of quorum sensing by RpoS in Pseudomonas aeruginosa. J. Bacteriol. 2000, 182, 4356–4360. [Google Scholar] [CrossRef] [Green Version]
- Rampioni, G.; Schuster, M.; Greenberg, E.P.; Bertani, I.; Grasso, M.; Venturi, V.; Zennaro, E.; Leoni, L. RsaL provides quorum sensing homeostasis and functions as a global regulator of gene expression in Pseudomonas aeruginosa. Mol. Microbiol. 2007, 66, 1557–1565. [Google Scholar] [CrossRef]
- Chugani, S.A.; Whiteley, M.; Lee, K.M.; D’Argenio, D.; Manoil, C.; Greenberg, E.P. QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2001, 98, 2752–2757. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Dong, Y.; Wu, D.; Bowler, M.W.; Zhang, L.; Song, H. QsIA disrupts LasR dimerization in antiactivation of bacterial quorum sensing. Proc. Natl. Acad. Sci. USA 2013, 110, 20765–20770. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Wang, Y.; Chen, F.; Xia, Y.; Lou, J.; Zhang, X.; Yang, N.; Sun, X.; Zhang, Q.; Zhuo, C.; et al. A Novel Signal Transduction Pathway that Modulates rhl Quorum Sensing and Bacterial Virulence in Pseudomonas aeruginosa. PLoS Pathog. 2014, 10, 8–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diggle, S.P.; Winzer, K.; Lazdunski, A.; Williams, P.; Cámara, M. Advancing the quorum in Pseudomonas aeruginosa: MvaT and the regulation of N-acylhomoserine lactone production and virulence gene expression. J. Bacteriol. 2002, 184, 2576–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, R.L.; Asfahl, K.L.; Van den Bossche, S.; Coenye, T.; Crabbé, A.; Dandekar, A.A. RhlR-regulated acyl-homoserine lactone quorum sensing in a cystic fibrosis isolate of Pseudomonas aeruginosa. MBio 2020, 11, e00532-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Aceves, M.P.; Cocotl-Yañez, M.; Servín-González, L.; Soberón-Chávez, G. The Rhl quorum-sensing system is at the top of the regulatory hierarchy under phosphate-limiting conditions in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2020, 203, e00475-20. [Google Scholar] [CrossRef]
- Heurlier, K.; Dénervaud, V.; Haas, D. Impact of quorum sensing on fitness of Pseudomonas aeruginosa. Int. J. Med. Microbiol. 2006, 296, 93–102. [Google Scholar] [CrossRef]
- Feltner, J.B.; Wolter, D.J.; Pope, C.E.; Groleau, M.C.; Smalley, N.E.; Greenberg, E.P.; Mayer-Hamblett, N.; Burns, J.; Deziel, E.; Hoffman, L.R.; et al. LasR Variant Cystic Fibrosis Isolates Reveal an Adaptable Quorum-Sensing Hierarchy in Pseudomonas aeruginosa. MBio 2016, 7, e01513-16. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Déziel, E.; Groleau, M.C.; Schaefer, A.L.; Greenberg, E.P. Social cheating in a Pseudomonas aeruginosa quorum-sensing variant. Proc. Natl. Acad. Sci. USA 2019, 116, 7021–7026. [Google Scholar] [CrossRef] [Green Version]
- Jensen, V.; Löns, D.; Zaoui, C.; Bredenbruch, F.; Meissner, A.; Dieterich, G.; Münch, R.; Häussler, S. RhlR expression in Pseudomonas aeruginosa is modulated by the Pseudomonas quinolone signal via PhoB-dependent and -independent pathways. J. Bacteriol. 2006, 188, 8601–8606. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Moustafa, D.A.; Stergioula, V.; Smith, C.D.; Goldberg, J.B.; Bassler, B.L. The PqsE and RhlR proteins are an autoinducer synthase-receptor pair that control virulence and biofilm development in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2018, 115, E9411–E9418. [Google Scholar] [CrossRef] [Green Version]
- Simanek, K.A.; Taylor, I.R.; Richael, E.K.; Lasek-Nesselquist, E.; Bassler, B.L.; Paczkowski, J.E. The PqsE-RhlR Interaction Regulates RhlR DNA Binding to Control Virulence Factor Production in Pseudomonas aeruginosa. Microbiol. Spectr. 2022, 10, e02108-21. [Google Scholar] [CrossRef]
- Taylor, I.R.; Paczkowski, J.E.; Jeffrey, P.D.; Henke, B.R.; Smith, C.D.; Bassler, B.L. Inhibitor Mimetic Mutations in the Pseudomonas aeruginosa PqsE Enzyme Reveal a Protein–Protein Interaction with the Quorum-Sensing Receptor RhlR That Is Vital for Virulence Factor Production. ACS Chem. Biol. 2021, 16, 740–752. [Google Scholar] [CrossRef] [PubMed]
- Zender, M.; Witzgall, F.; Drees, S.L.; Weidel, E.; Maurer, C.K.; Fetzner, S.; Blankenfeldt, W.; Empting, M.; Hartmann, R.W. Dissecting the Multiple Roles of PqsE in Pseudomonas aeruginosa Virulence by Discovery of Small Tool Compounds. ACS Chem. Biol. 2016, 11, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Farrow, J.M., 3rd; Sund, Z.M.; Ellison, M.L.; Wade, D.S.; Coleman, J.P.; Pesci, E.C. PqsE functions independently of PqsR-Pseudomonas quinolone signal and enhances the rhl quorum-sensing system. J. Bacteriol. 2008, 190, 7043–7051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa lifestyle: A paradigm for adaptation, survival, and persistence. Front. Cell. Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Limoli, D.H.; Hoffman, L.R. Help, hinder, hide and harm: What can we learn from the interactions between Pseudomonas aeruginosa and Staphylococcus aureus during respiratory infections. Thorax 2019, 74, 684–692. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Obata, K.; Keira, T.; Miyata, R.; Hirakawa, S.; Takano, K.; Kohno, T.; Sawada, N.; Himi, T.; Kojima, T. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir. Res. 2014, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.B.; DiMango, E.; Bryan, R.; Gambello, M.; Iglewski, B.H.; Goldberg, J.B.; Prince, A. Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect. Immun. 1996, 64, 37–43. Available online: https://www.ncbi.nlm.nih.gov/pubmed/8557368 (accessed on 25 November 2021). [CrossRef] [Green Version]
- Parmely, M.; Gale, A.; Clabaugh, M.; Horvat, R.; Zhou, W.W. Proteolytic inactivation of cytokines by Pseudomonas aeruginosa. Infect. Immun. 1990, 58, 3009–3014. [Google Scholar] [CrossRef] [Green Version]
- LaFayette, S.L.; Houle, D.; Beaudoin, T.; Wojewodka, G.; Radzioch, D.; Hoffman, L.R.; Burns, J.L.; Dandekar, A.A.; Smalley, N.E.; Chandler, J.R.; et al. Cystic fibrosis–adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci. Adv. 2015, 1, e1500199. [Google Scholar] [CrossRef] [Green Version]
- Casilag, F.; Lorenz, A.; Krueger, J.; Klawonn, F.; Weiss, S.; Häussler, S. LasB elastase of Pseudomonas aeruginosa acts in concert with alkaline protease AprA to prevent flagellin-mediated immune recognition. Infect. Immun. 2015, 84, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Lozano, C.; Azcona-Gutiérrez, J.M.; Van Bambeke, F.; Sáenz, Y. Great phenotypic and genetic variation among successive chronic Pseudomonas aeruginosa from a cystic fibrosis patient. PLoS ONE 2018, 13, e0204167. [Google Scholar] [CrossRef] [PubMed]
- Zupetic, J.; Peñaloza, H.F.; Bain, W.; Hulver, M.; Mettus, R.; Jorth, P.; Doi, Y.; Bomberger, J.; Pilewski, J.; Nouraie, M.; et al. Elastase Activity From Pseudomonas aeruginosa Respiratory Isolates and ICU Mortality. Chest 2021, 160, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Tuli, J.F.; Ramezanpour, M.; Cooksley, C.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Association between mucosal barrier disruption by Pseudomonas aeruginosa exoproteins and asthma in patients with chronic rhinosinusitis. Allergy Eur. J. Allergy Clin. Immunol. 2021, 76, 3459–3469. [Google Scholar] [CrossRef] [PubMed]
- Rampioni, G.; Schuster, M.; Greenberg, E.P.; Zennaro, E.; Leoni, L. Contribution of the RsaL global regulator to Pseudomonas aeruginosa virulence and biofilm formation. FEMS Microbiol. Lett. 2009, 301, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, L.E.; Price-Whelan, A.; Petersen, A.; Whiteley, M.; Newman, D.K. The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol. Microbiol. 2006, 61, 1308–1321. [Google Scholar] [CrossRef]
- Cocotl-Yañez, M.; Soto-Aceves, M.P.; González-Valdez, A.; Servín-González, L.; Soberón-Chávez, G. Virulence factors regulation by the quorum-sensing and Rsm systems in the marine strain Pseudomonas aeruginosa ID4365, a natural mutant in lasR. FEMS Microbiol. Lett. 2021, 367, fnaa092. [Google Scholar] [CrossRef]
- Hall, S.; McDermott, C.; Anoopkumar-Dukie, S.; McFarland, A.J.; Forbes, A.; Perkins, A.V.; Davey, A.K.; Chess-Williams, R.; Kiefel, M.J.; Arora, D.; et al. Cellular effects of pyocyanin, a secreted virulence factor of Pseudomonas aeruginosa. Toxins. 2016, 8, 236. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.; Williams, D.; Fothergill, J.L.; Paterson, S.; Winstanley, C.; Brockhurst, M.A. High virulence sub-populations in Pseudomonas aeruginosa long-term cystic fibrosis airway infections. BMC Microbiol. 2017, 17, 30. [Google Scholar] [CrossRef] [Green Version]
- Gupte, A.; Jyot, J.; Ravi, M.; Ramphal, R. High pyocyanin production and non-motility of Pseudomonas aeruginosa isolates are correlated with septic shock or death in bacteremic patients. PLoS ONE 2021, 16, e0253259. [Google Scholar] [CrossRef]
- Meirelles, L.A.; Newman, D.K. Both toxic and beneficial effects of pyocyanin contribute to the lifecycle of Pseudomonas aeruginosa. Mol. Microbiol. 2018, 110, 995–1010. [Google Scholar] [CrossRef] [Green Version]
- Saunders, S.H.; Tse, E.C.M.; Yates, M.D.; Otero, F.J.; Trammell, S.A.; Stemp, E.D.A.; Barton, J.K.; Tender, L.M.; Newman, D.K. Extracellular DNA Promotes Efficient Extracellular Electron Transfer by Pyocyanin in Pseudomonas aeruginosa Biofilms. Cell 2020, 182, 919–932.e19. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.; Dockrell, D.H.; Pattery, T.; Lee, D.G.; Cornelis, P.; Hellewell, P.G.; Whyte, M.K.B. Pyocyanin Production by Pseudomonas aeruginosa Induces Neutrophil Apoptosis and Impairs Neutrophil-Mediated Host Defenses In Vivo. J. Immunol. 2005, 174, 3643–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.; Manefield, M. Pyocyanin Promotes Extracellular DNA Release in Pseudomonas aeruginosa. PLoS ONE 2012, 7, e46718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caçador, N.C.; Paulino da Costa Capizzani, C.; Monteiro Marin Torres, L.A.G.; Galetti, R.; Ciofu, O.; Lúcia da Costa Darini, A.; Høiby, N. Adaptation of Pseudomonas aeruginosa to the chronic phenotype by mutations in the algTmucABD operon in isolates from Brazilian cystic fibrosis patients. PLoS ONE 2018, 13, e0208013. [Google Scholar] [CrossRef]
- Stapper, A.P.; Narasimhan, G.; Ohman, D.E.; Barakat, J.; Hentzer, M.; Molin, S.; Kharazmi, A.; Høiby, N.; Mathee, K. Alginate production affects Pseudomonas aeruginosa biofilm development and architecture, but is not essential for biofilm formation. J. Med. Microbiol. 2004, 53, 679–690. [Google Scholar] [CrossRef]
- Ciofu, O.; Mandsberg, L.F.; Bjarnsholt, T.; Wassermann, T.; Høiby, N. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: Strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 2010, 156, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Cross, A.R.; Raghuram, V.; Wang, Z.; Dey, D.; Goldberg, J.B. Overproduction of the AlgT sigma factor is lethal to mucoid Pseudomonas aeruginosa. J. Bacteriol. 2020, 202, e00445-20. [Google Scholar] [CrossRef]
- Harrison, J.J.; Almblad, H.; Irie, Y.; Wolter, D.J.; Eggleston, H.C.; Randall, T.E.; Kitzman, J.O.; Stackhouse, B.; Emerson, J.C.; McNamara, S.; et al. Elevated exopolysaccharide levels in Pseudomonas aeruginosa flagellar mutants have implications for biofilm growth and chronic infections. PLoS Genet. 2020, 16, e1008848. [Google Scholar] [CrossRef]
- Jennings, L.K.; Dreifus, J.E.; Reichhardt, C.; Storek, K.M.; Secor, P.R.; Wozniak, D.J.; Hisert, K.B.; Parsek, M.R. Pseudomonas aeruginosa aggregates in cystic fibrosis sputum produce exopolysaccharides that likely impede current therapies. Cell Rep. 2021, 34, 108782. [Google Scholar] [CrossRef]
- Reichhardt, C.; Jacobs, H.M.; Matwichuk, M.; Wong, C.; Wozniak, D.J.; Parsek, M.R. The Versatile Pseudomonas aeruginosa Biofilm Matrix Protein CdrA Promotes Aggregation through Different Extracellular Exopolysaccharide Interactions. J. Bacteriol. 2020, 202, e00216-20. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Schiessl, K.T.; Hu, F.; Jo, J.; Nazia, S.Z.; Wang, B.; Price-Whelan, A.; Min, W.; Dietrich, L.E.P. Phenazine production promotes antibiotic tolerance and metabolic heterogeneity in Pseudomonas aeruginosa biofilms. Nat. Commun. 2019, 10, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Wei, Q.; Zhao, T.; Guo, Y.; Ma, L.Z. A survival strategy for Pseudomonas aeruginosa that uses exopolysaccharides to sequester and store iron to stimulate psl-dependent biofilm formation. Appl. Environ. Microbiol. 2016, 82, 6403–6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichhardt, C.; Wong, C.; da Silva, D.P.; Wozniak, D.J.; Parsek, M.R. CDRA interactions within the Pseudomonas aeruginosa biofilm matrix safeguard it from proteolysis and promote cellular packing. MBio 2018, 9, e01376-18. [Google Scholar] [CrossRef] [Green Version]
- Passos da Silva, D.; Matwichuk, M.L.; Townsend, D.O.; Reichhardt, C.; Lamba, D.; Wozniak, D.J.; Parsek, M.R. The Pseudomonas aeruginosa lectin LecB binds to the exopolysaccharide Psl and stabilizes the biofilm matrix. Nat. Commun. 2019, 10, 2183. [Google Scholar] [CrossRef]
- Gutiérrez-Gómez, U.; Soto-Aceves, M.P.; Servín-González, L.; Soberón-Chávez, G. Overproduction of rhamnolipids in Pseudomonas aeruginosa PA14 by redirection of the carbon flux from polyhydroxyalkanoate synthesis and overexpression of the rhlAB-R operon. Biotechnol. Lett. 2018, 40, 1561–1566. [Google Scholar] [CrossRef]
- Pessi, G.; Haas, D. Transcriptional control of the hydrogen cyanide biosynthetic genes hcnABC by the anaerobic regulator ANR and the quorum-sensing regulators LasR and RhlR in Pseudomonas aeruginosa. J. Bacteriol. 2000, 182, 6940–6949. [Google Scholar] [CrossRef] [Green Version]
- Cody, W.L.; Pritchett, C.L.; Jones, A.K.; Carterson, A.J.; Jackson, D.; Frisk, A.; Wolfgang, M.C.; Schurr, M.J. Pseudomonas aeruginosa AlgR controls cyanide production in an AlgZ-dependent manner. J. Bacteriol. 2009, 191, 2993–3002. [Google Scholar] [CrossRef] [Green Version]
- Blier, A.S.; Vieillard, J.; Gerault, E.; Dagorn, A.; Varacavoudin, T.; Le Derf, F.; Orange, N.; Feuilloley, M.; Lesouhaitier, O. Quantification of Pseudomonas aeruginosa hydrogen cyanide production by a polarographic approach. J. Microbiol. Methods 2012, 90, 20–24. [Google Scholar] [CrossRef]
- Enderby, B.; Smith, D.; Carroll, W.; Lenney, W. Hydrogen cyanide as a biomarker for Pseudomonas aeruginosa in the breath of children with cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 142–147. [Google Scholar] [CrossRef]
- Smith, D.; Španěl, P.; Gilchrist, F.J.; Lenney, W. Hydrogen cyanide, a volatile biomarker of Pseudomonas aeruginosa infection. J. Breath Res. 2013, 7, 044001. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Asfahl, K.L.; Li, N.; Sun, F.; Xiao, J.; Shen, D.; Dandekar, A.A.; Wang, M. Conditional quorum-sensing induction of a cyanide-insensitive terminal oxidase stabilizes cooperating populations of Pseudomonas aeruginosa. Nat. Commun. 2019, 10, 4999. [Google Scholar] [CrossRef] [PubMed]
- Darch, S.E.; McNally, A.; Harrison, F.; Corander, J.; Barr, H.L.; Paszkiewicz, K.; Holden, S.; Fogarty, A.; Crusz, S.A.; Diggle, S.P. Recombination is a key driver of genomic and phenotypic diversity in a Pseudomonas aeruginosa population during cystic fibrosis infection. Sci. Rep. 2015, 5, 7649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandeplassche, E.; Sass, A.; Lemarcq, A.; Dandekar, A.A.; Coenye, T.; Crabbé, A. In vitro evolution of Pseudomonas aeruginosa AA2 biofilms in the presence of cystic fibrosis lung microbiome members. Sci. Rep. 2019, 9, 12859. [Google Scholar] [CrossRef] [Green Version]
- Acosta, N.; Waddell, B.; Heirali, A.; Somayaji, R.; Surette, M.G.; Workentine, M.L.; Rabin, H.R.; Parkins, M.D. Cystic Fibrosis Patients Infected With Epidemic Pseudomonas aeruginosa Strains Have Unique Microbial Communities. Front. Cell. Infect. Microbiol. 2020, 10, 173. [Google Scholar] [CrossRef]
- Faure, E.; Kwong, K.; Nguyen, D. Pseudomonas aeruginosa in Chronic Lung Infections: How to Adapt Within the Host? Front. Immunol. 2018, 9, 2416. [Google Scholar] [CrossRef] [Green Version]
- Mould, D.L.; Botelho, N.J.; Hogan, D.A. Intraspecies signaling between common variants of Pseudomonas aeruginosa increases production of quorum-sensing-controlled virulence factors. MBio 2020, 11, e01865-20. [Google Scholar] [CrossRef]
- Vipin, C.; Mujeeburahiman, M.; Arun, A.B.; Ashwini, P.; Mangesh, S.V.; Rekha, P.D. Adaptation and diversification in virulence factors among urinary catheter-associated Pseudomonas aeruginosa isolates. J. Appl. Microbiol. 2019, 126, 641–650. [Google Scholar] [CrossRef]
- Jeske, A.; Arce-Rodriguez, A.; Thöming, J.G.; Tomasch, J.; Häussler, S. Evolution of biofilm-adapted gene expression profiles in lasR-deficient clinical Pseudomonas aeruginosa isolates. NPJ Biofilms Microbiomes 2022, 8, 6. [Google Scholar] [CrossRef]
- Bartell, J.A.; Sommer, L.M.; Marvig, R.L.; Skov, M.; Pressler, T.; Molin, S.; Johansen, H.K. Omics-based tracking of Pseudomonas aeruginosa persistence in “eradicated” cystic fibrosis patients. Eur. Respir. J. 2021, 57, 2000512. [Google Scholar] [CrossRef]
- Gonzalez, M.R.; Ducret, V.; Leoni, S.; Fleuchot, B.; Jafari, P.; Raffoul, W.; Applegate, L.A.; Que, Y.A.; Perron, K. Transcriptome analysis of Pseudomonas aeruginosa cultured in human burn wound exudates. Front. Cell. Infect. Microbiol. 2018, 8, 39. [Google Scholar] [CrossRef]
- Kumar, R.; Chhibber, S.; Harjai, K. Quorum sensing is necessary for the virulence of Pseudomonas aeruginosa during urinary tract infection. Kidney Int. 2009, 76, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricks-Lima, J.; Hendrickson, C.M.; Allgaier, M.; Zhuo, H.; Wiener-Kronish, J.P.; Lynch, S.V.; Yang, K. Differences in biofilm formation and antimicrobial resistance of Pseudomonas aeruginosa isolated from airways of mechanically ventilated patients and cystic fibrosis patients. Int. J. Antimicrob. Agents 2011, 37, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Ugwuanyi, F.C.; Ajayi, A.; Ojo, D.A.; Adeleye, A.I.; Smith, S.I. Evaluation of efflux pump activity and biofilm formation in multidrug resistant clinical isolates of Pseudomonas aeruginosa isolated from a Federal Medical Center in Nigeria. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 11. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.H.; Kwon, K.C.; Kim, S.; Park, Y.; Koo, S.H. Association between biofilm formation and antimicrobial resistance in carbapenem-resistant Pseudomonas aeruginosa. Ann. Clin. Lab. Sci. 2018, 48, 363–368. [Google Scholar] [PubMed]
- Ahmed, M.N.; Porse, A.; Sommer, M.O.A.; Høiby, N.; Ciofu, O. Evolution of antibiotic resistance in biofilm and planktonic Pseudomonas aeruginosa populations exposed to subinhibitory levels of ciprofloxacin. Antimicrob. Agents Chemother. 2018, 62, e00320-18. [Google Scholar] [CrossRef] [Green Version]
- Soh, E.Y.C.; Smith, F.; Gimenez, M.R.; Yang, L.; Vejborg, R.M.; Fletcher, M.; Halliday, N.; Bleves, S.; Heeb, S.; Cámara, M.; et al. Disruption of the Pseudomonas aeruginosa Tat system perturbs PQS-dependent quorum sensing and biofilm maturation through lack of the Rieske cytochrome bc1sub-unit. PLoS Pathog. 2021, 17, e1009425. [Google Scholar] [CrossRef]
- Allesen-Holm, M.; Barken, K.B.; Yang, L.; Klausen, M.; Webb, J.S.; Kjelleberg, S.; Molin, S.; Givskov, M.; Tolker-Nielsen, T. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 2006, 59, 1114–1128. [Google Scholar] [CrossRef]
- Chiang, W.C.; Nilsson, M.; Jensen, P.Ø.; Høiby, N.; Nielsen, T.E.; Givskov, M.; Tolker-Nielsen, T. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2013, 57, 2352–2361. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef]
- Schertzer, J.W.; Whiteley, M. A bilayer-couple model of bacterial outer membrane vesicle biogenesis. MBio 2012, 3, e00297-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessel, A.K.; Liew, J.; Kwon, T.; Marcotte, E.M.; Whiteley, M. Role of Pseudomonas aeruginosa peptidoglycan-associated outer membrane proteins in vesicle formation. J. Bacteriol. 2013, 195, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussouni, M.; Berry, L.; Sipka, T.; Nguyen-Chi, M.; Blanc-Potard, A.B. Pseudomonas aeruginosa OprF plays a role in resistance to macrophage clearance during acute infection. Sci. Rep. 2021, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Fito-Boncompte, L.; Chapalain, A.; Bouffartigues, E.; Chaker, H.; Lesouhaitier, O.; Gicquel, G.; Bazire, A.; Madi, A.; Connil, N.; Véron, W.; et al. Full virulence of Pseudomonas aeruginosa requires OprF. Infect. Immun. 2011, 79, 1176–1186. [Google Scholar] [CrossRef] [Green Version]
- Manning, A.J.; Kuehn, M.J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 2011, 11, 258. [Google Scholar] [CrossRef] [Green Version]
- Bouffartigues, E.; Moscoso, J.A.; Duchesne, R.; Rosay, T.; Fito-Boncompte, L.; Gicquel, G.; Maillot, O.; Bénard, M.; Bazire, A.; Brenner-Weiss, G.; et al. The absence of the Pseudomonas aeruginosa OprF protein leads to increased biofilm formation through variation in c-di-GMP level. Front. Microbiol. 2015, 6, 630. [Google Scholar] [CrossRef] [Green Version]
- Hassett, D.J.; Cuppoletti, J.; Trapnell, B.; Lymar, S.V.; Rowe, J.J.; Yoon, S.S.; Hilliard, G.M.; Parvatiyar, K.; Kamani, M.C.; Wozniak, D.J.; et al. Anaerobic metabolism and quorum sensing by Pseudomonas aeruginosa biofilms in chronically infected cystic fibrosis airways: Rethinking antibiotic treatment strategies and drug targets. Adv. Drug Deliv. Rev. 2002, 54, 1425–1443. [Google Scholar] [CrossRef]
- Turner, K.H.; Everett, J.; Trivedi, U.; Rumbaugh, K.P.; Whiteley, M. Requirements for Pseudomonas aeruginosa Acute Burn and Chronic Surgical Wound Infection. PLoS Genet. 2014, 10, e1004518. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.S.; Hennigan, R.F.; Hilliard, G.M.; Ochsner, U.A.; Parvatiyar, K.; Kamani, M.C.; Allen, H.L.; DeKievit, T.R.; Gardner, P.R.; Schwab, U.; et al. Pseudomonas aeruginosa anaerobic respiration in biofilms: Relationships to cystic fibrosis pathogenesis. Dev. Cell 2002, 3, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Garai, P.; Berry, L.; Moussouni, M.; Bleves, S.; Blanc-Potard, A.B. Killing from the inside: Intracellular role of T3SS in the fate of Pseudomonas aeruginosa within macrophages revealed by mgtC and oprF mutants. PLoS Pathog. 2019, 15, e1007812. [Google Scholar] [CrossRef] [Green Version]
- Ude, J.; Tripathi, V.; Buyck, J.M.; Söderholm, S.; Cunrath, O.; Fanous, J.; Claudi, B.; Egli, A.; Schleberger, C.; Hiller, S.; et al. Outer membrane permeability: Antimicrobials and diverse nutrients bypass porins in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2021, 118, e2107644118. [Google Scholar] [CrossRef] [PubMed]
- Richardot, C.; Plésiat, P.; Fournier, D.; Monlezun, L.; Broutin, I.; Llanes, C. Carbapenem resistance in cystic fibrosis strains of Pseudomonas aeruginosa as a result of amino acid substitutions in porin OprD. Int. J. Antimicrob. Agents 2015, 45, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Kos, V.N.; McLaughlin, R.E.; Gardner, H.A. Identification of unique in-frame deletions in OprD among clinical isolates of Pseudomonas aeruginosa. Pathog. Dis. 2016, 74, ftw031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiani, M.; Astani, A.; Eslami, G.; Khaledi, M.; Afkhami, H.; Rostami, S.; Zarei, M.; Rezaei Khozani, N.; Zandi, H. Upstream region of OprD mutations in imipenem-resistant and imipenem-sensitive Pseudomonas isolates. AMB Express 2021, 11, 82. [Google Scholar] [CrossRef]
- Atrissi, J.; Milan, A.; Bressan, R.; Lucafò, M.; Petix, V.; Busetti, M.; Dolzani, L.; Lagatolla, C. Interplay of OpdP Porin and Chromosomal Carbapenemases in the Determination of Carbapenem Resistance/Susceptibility in Pseudomonas aeruginosa. Microbiol. Spectr. 2021, 9, e01186-21. [Google Scholar] [CrossRef]
- Sonnleitner, E.; Pusic, P.; Wolfinger, M.T.; Bläsi, U. Distinctive Regulation of Carbapenem Susceptibility in Pseudomonas aeruginosa by Hfq. Front. Microbiol. 2020, 11, 1001. [Google Scholar] [CrossRef]
- Raavi; Mishra, S.; Singh, S. Prevention of OprD regulated antibiotic resistance in Pseudomonas aeruginosa biofilm. Microb. Pathog. 2017, 112, 221–229. [Google Scholar] [CrossRef]
- Dieppois, G.; Ducret, V.; Caille, O.; Perron, K. The transcriptional regulator CzcR modulates antibiotic resistance and quorum sensing in Pseudomonas aeruginosa. PLoS ONE 2012, 7, e38148. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Ding, S.; Chen, F.; Zhang, X.; Xia, Y.; Di, H.; Cao, Q.; Deng, X.; Wu, M.; Wong, C.C.L.; et al. The Crc protein participates in down-regulation of the Lon gene to promote rhamnolipid production and rhl quorum sensing in Pseudomonas aeruginosa. Mol. Microbiol. 2015, 96, 526–547. [Google Scholar] [CrossRef]
- Amsalu, A.; Sapula, S.A.; Lopes, M.D.B.; Hart, B.J.; Nguyen, A.H.; Drigo, B.; Turnidge, J.; Leong, L.E.X.; Venter, H. Efflux pump-driven antibiotic and biocide cross-resistance in Pseudomonas aeruginosa isolated from different ecological Niches: A case study in the development of multidrug resistance in environmental hotspots. Microorganisms 2020, 8, 1647. [Google Scholar] [CrossRef]
- Ma, Z.; Xu, C.; Zhang, X.; Wang, D.; Pan, X.; Liu, H.; Zhu, G.; Bai, F.; Cheng, Z.; Wu, W.; et al. A MexR Mutation Which Confers Aztreonam Resistance to Pseudomonas aeruginosa. Front. Microbiol. 2021, 12, 1503. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Ghosh, A.; Chanda, D.D.; Das Talukdar, A.; Choudhury, M.D.; Paul, D.; Maurya, A.P.; Chakravorty, A.; Bhattacharjee, A. Premature termination of MexR leads to overexpression of MexAB-OprM efflux pump in Pseudomonas aeruginosa in a tertiary referral hospital in India. PLoS ONE 2016, 11, e0149156. [Google Scholar]
- Tian, Z.X.; Yi, X.X.; Cho, A.; O’Gara, F.; Wang, Y.P. CpxR Activates MexAB-OprM Efflux Pump Expression and Enhances Antibiotic Resistance in Both Laboratory and Clinical nalB-Type Isolates of Pseudomonas aeruginosa. PLoS Pathog. 2016, 12, e1005932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minagawa, S.; Inami, H.; Kato, T.; Sawada, S.; Yasuki, T.; Miyairi, S.; Horikawa, M.; Okuda, J.; Gotoh, N. RND type efflux pump system MexAB-OprM of Pseudomonas aeruginosa selects bacterial languages, 3-oxo-acyl-homoserine lactones, for cell-to-cell communication. BMC Microbiol. 2012, 12, 70. [Google Scholar] [CrossRef] [Green Version]
- Maseda, H.; Sawada, I.; Saito, K.; Uchiyama, H.; Nakae, T.; Nomura, N. Enhancement of the mexAB-oprM Efflux Pump Expression by a Quorum-Sensing Autoinducer and Its Cancellation by a Regulator, MexT, of the mexEF-oprN Efflux Pump Operon in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Horna, G.; López, M.; Guerra, H.; Saénz, Y.; Ruiz, J. Interplay between MexAB-OprM and MexEF-OprN in clinical isolates of Pseudomonas aeruginosa. Sci. Rep. 2018, 8, 16463. [Google Scholar] [CrossRef]
- Llanes, C.; Köhler, T.; Patry, I.; Dehecq, B.; Van Delden, C.; Plésiat, P. Role of the MexEF-OprN efflux system in low-level resistance of Pseudomonas aeruginosa to ciprofloxacin. Antimicrob. Agents Chemother. 2011, 55, 5676–5684. [Google Scholar] [CrossRef] [Green Version]
- Juarez, P.; Broutin, I.; Bordi, C.; Plésiat, P.; Llanes, C. Constitutive activation of MexT by amino acid substitutions results in MexEF-OprN overproduction in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e02445-17. [Google Scholar] [CrossRef] [Green Version]
- Richardot, C.; Juarez, P.; Jeannot, K.; Patry, I.; Plésiat, P.; Llanes, C. Amino acid substitutions account for most mexS alterations in clinical nfxC mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 2302–2310. [Google Scholar] [CrossRef] [Green Version]
- Smalley, N.E.; An, D.; Parsek, M.R.; Chandler, J.R.; Dandekar, A.A. Quorum sensing protects Pseudomonas aeruginosa against cheating by other species in a laboratory coculture model. J. Bacteriol. 2015, 197, 3154–3159. [Google Scholar] [CrossRef] [Green Version]
- Boyle, K.E.; Monaco, H.; van Ditmarsch, D.; Deforet, M.; Xavier, J.B. Integration of Metabolic and Quorum Sensing Signals Governing the Decision to Cooperate in a Bacterial Social Trait. PLoS Comput. Biol. 2015, 11, e1004279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshri, R.D.; Zrihen, K.S.; Shner, I.; Omer Bendori, S.; Eldar, A. Selection for increased quorum-sensing cooperation in Pseudomonas aeruginosa through the shut-down of a drug resistance pump. ISME J. 2018, 12, 2458–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, K.C.; Benomar, S.; Camuy-Vélez, L.A.; Nasseri, E.B.; Wang, X.; Neuenswander, B.; Chandler, J.R. Quorum-sensing control of antibiotic resistance stabilizes cooperation in Chromobacterium violaceum. ISME J. 2018, 12, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Poole, K.; Gotoh, N.; Tsujimoto, H.; Zhao, Q.; Wada, A.; Yamasaki, T.; Neshat, S.; Yamagishi, J.; Li, X.-Z.; Nishino, T. Overexpression of the mexC–mexD–oprJ efflux operon in nfxB-type multidrug-resistant strains of Pseudomonas aeruginosa. Mol. Microbiol. 1996, 21, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Alcalde-Rico, M.; Olivares-Pacheco, J.; Alvarez-Ortega, C.; Cámara, M.; Martínez, J.L. Role of the multidrug resistance efflux pump MexCD-OprJ in the Pseudomonas aeruginosa quorum sensing response. Front. Microbiol. 2018, 9, 2752. [Google Scholar] [CrossRef] [PubMed]
- Déziel, E.; Gopalan, S.; Tampakaki, A.P.; Lépine, F.; Padfield, K.E.; Saucier, M.; Xiao, G.; Rahme, L.G. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: Multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl- l-homoserine lactones. Mol. Microbiol. 2005, 55, 998–1014. [Google Scholar] [CrossRef]
- Hodgkinson, J.T.; Gross, J.; Baker, Y.R.; Spring, D.R.; Welch, M. A new Pseudomonas quinolone signal (PQS) binding partner: MexG. Chem. Sci. 2016, 7, 2553–2562. [Google Scholar] [CrossRef] [Green Version]
- Sakhtah, H.; Koyama, L.; Zhang, Y.; Morales, D.K.; Fields, B.L.; Price-Whelan, A.; Hogan, D.A.; Shepard, K.; Dietrich, L.E.P. The Pseudomonas aeruginosa efflux pump MexGHI-OpmD transports a natural phenazine that controls gene expression and biofilm development. Proc. Natl. Acad. Sci. USA 2016, 113, E3538–E3547. [Google Scholar] [CrossRef] [Green Version]
- Welsh, M.A.; Eibergen, N.R.; Moore, J.D.; Blackwell, H.E. Small molecule disruption of quorum sensing cross-regulation in Pseudomonas aeruginosa causes major and unexpected alterations to virulence phenotypes. J. Am. Chem. Soc. 2015, 137, 1510–1519. [Google Scholar] [CrossRef] [Green Version]
- Borlee, B.R.; Geske, G.D.; Blackwell, H.E.; Handelsman, J. Identification of synthetic inducers and inhibitors of the quorum-sensing regulator LasR in Pseudomonas aeruginosa by high-throughput screening. Appl. Env. Microbiol. 2010, 76, 8255–8258. [Google Scholar] [CrossRef] [Green Version]
- Bjarnsholt, T.; Tolker-Nielsen, T.; Hoiby, N.; Givskov, M. Interference of Pseudomonas aeruginosa signalling and biofilm formation for infection control. Expert Rev. Mol. Med. 2010, 12, e11. [Google Scholar] [CrossRef] [PubMed]
- Paczkowski, J.E.; Mukherjee, S.; McCready, A.R.; Cong, J.-P.; Aquino, C.J.; Kim, H.; Henke, B.R.; Smith, C.D.; Bassler, B.L. Flavonoids suppress Pseudomonas aeruginosa virulence through allosteric inhibition of quorum-sensing Receptors. J. Biol. Chem. 2017, 292, 4064–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldelli, V.; D’Angelo, F.; Pavoncello, V.; Fiscarelli, E.V.; Visca, P.; Rampioni, G.; Leoni, L. Identification of FDA-approved antivirulence drugs targeting the Pseudomonas aeruginosa quorum sensing effector protein PqsE. Virulence 2020, 11, 652–668. [Google Scholar] [CrossRef] [PubMed]
- Gerdt, J.P.; McInnis, C.E.; Schell, T.L.; Rossi, F.M.; Blackwell, H.E. Mutational analysis of the quorum-sensing receptor LasR reveals interactions that govern activation and inhibition by nonlactone ligands. Chem. Biol. 2014, 21, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, M.J.; Muraglia, E.; Bazzo, R.; Carfi, A. Molecular insights into quorum sensing in the human pathogen Pseudomonas aeruginosa from the structure of the virulence regulator LasR bound to its autoinducer. J. Biol. Chem. 2007, 282, 13592–13600. [Google Scholar] [CrossRef] [Green Version]
- Paczkowski, J.E.; McCready, A.R.; Cong, J.-P.; Li, Z.; Jeffrey, P.D.; Smith, C.D.; Henke, B.R.; Hughson, F.M.; Bassler, B.L. An Autoinducer Analogue Reveals an Alternative Mode of Ligand Binding for the LasR Quorum-Sensing Receptor. ACS Chem. Biol. 2019, 14, 378–389. [Google Scholar] [CrossRef]
- McCready, A.R.; Paczkowski, J.E.; Henke, B.R.; Bassler, B.L. Structural determinants driving homoserine lactone ligand selection in the Pseudomonas aeruginosa LasR quorum-sensing receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Swem, L.R.; Swem, D.L.; Stauff, D.L.; O’Loughlin, C.T.; Jeffrey, P.D.; Bassler, B.L.; Hughson, F.M. A strategy for antagonizing quorum sensing. Mol. Cell 2011, 42, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Cafora, M.; Deflorian, G.; Forti, F.; Ferrari, L.; Binelli, G.; Briani, F.; Ghisotti, D.; Pistocchi, A. Phage therapy against Pseudomonas aeruginosa infections in a cystic fibrosis zebrafish model. Sci. Rep. 2019, 9, 1527. [Google Scholar] [CrossRef] [Green Version]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Aslam, S.; Courtwright, A.M.; Koval, C.; Lehman, S.M.; Morales, S.; Furr, C.L.L.; Rosas, F.; Brownstein, M.J.; Fackler, J.R.; Sisson, B.M.; et al. Early clinical experience of bacteriophage therapy in 3 lung transplant recipients. Am. J. Transplant. 2019, 19, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Law, N.; Logan, C.; Yung, G.; Furr, C.L.L.; Lehman, S.M.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; et al. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection 2019, 47, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Stanley, G.; Modak, M.; Koff, J.L.; Turner, P.E. Bacteriophage therapy for infections in CF. Pediatr. Pulmonol. 2021, 56, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Hoyland-Kroghsbo, N.M.; Paczkowski, J.; Mukherjee, S.; Broniewski, J.; Westra, E.; Bondy-Denomy, J.; Bassler, B.L.L.; Høyland-Kroghsbo, N.M.; Paczkowski, J.; Mukherjee, S.; et al. Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci. USA 2017, 114, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Patterson, A.G.; Jackson, S.A.; Taylor, C.; Evans, G.B.; Salmond, G.P.C.; Przybilski, R.; Staals, R.H.J.; Fineran, P.C. Quorum Sensing Controls Adaptive Immunity through the Regulation of Multiple CRISPR-Cas Systems. Mol. Cell 2016, 64, 1102–1108. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, T.C.; Cywes-Bentley, C.; Moeller, T.D.; Weyant, K.B.; Putnam, D.; Chang, Y.F.; Jones, B.D.; Pier, G.B.; DeLisa, M.P. Immunization with outer membrane vesicles displaying conserved surface polysaccharide antigen elicits broadly antimicrobial antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, E3106–E3115. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, M.; Behrouz, B.; Irajian, G.; Mahdavi, M.; Korpi, F.; Motamedifar, M. Passive immunization against Pseudomonas aeruginosa recombinant PilA in a murine burn wound model. Microb. Pathog. 2016, 101, 83–88. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Class | Mechanism of Action | Total Resistance (%) | Isolates Tested | Trend | HAI Type with Highest% Resistance |
|---|---|---|---|---|---|---|
| Imipenem, meropenem | Carbapenem | Peptidoglycan crosslinking/cell wall synthesis | 13.3% | 6665 | −1% | CLABSI |
| Gentamicin, tobramycin, amikacin, streptomycin | Aminoglycoside | Protein synthesis | 8.5% | 7931 | −0.6% | CAUTI |
| Ciprofloxacin, levofloxacin | Fluoroquinolone | Topoisomerase/DNA synthesis | 15.2% | 7795 | −1.6% | CAUTI |
| Cefazolin, cefepime, ceftriaxone, ceftazidime | Cephalosporin | Peptidoglycan/cell wall synthesis | 15.1% | 7879 | −0.9% | CLABSI/CAUTI |
| Piperacillin, tazobactam | Penicillin | Peptidoglycan crosslinking/cell wall synthesis | 11.9% | 7467 | −0.1% | CLABSI/CAUTI |
| Multi-drug | 7.9% | 7940 | −2.1% | CAUTI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simanek, K.A.; Paczkowski, J.E. Resistance Is Not Futile: The Role of Quorum Sensing Plasticity in Pseudomonas aeruginosa Infections and Its Link to Intrinsic Mechanisms of Antibiotic Resistance. Microorganisms 2022, 10, 1247. https://doi.org/10.3390/microorganisms10061247
Simanek KA, Paczkowski JE. Resistance Is Not Futile: The Role of Quorum Sensing Plasticity in Pseudomonas aeruginosa Infections and Its Link to Intrinsic Mechanisms of Antibiotic Resistance. Microorganisms. 2022; 10(6):1247. https://doi.org/10.3390/microorganisms10061247
Chicago/Turabian StyleSimanek, Kayla A., and Jon E. Paczkowski. 2022. "Resistance Is Not Futile: The Role of Quorum Sensing Plasticity in Pseudomonas aeruginosa Infections and Its Link to Intrinsic Mechanisms of Antibiotic Resistance" Microorganisms 10, no. 6: 1247. https://doi.org/10.3390/microorganisms10061247