
Environmental Selection Shapes Bacterial Community Composition in Traditionally Fermented Maize-Based Foods from Benin, Tanzania and Zambia

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction and Sequencing
2.2. Data Processing
2.2.1. Sequence-Quality, Denoising and Dereplication
2.2.2. Taxonomic Classification
2.2.3. Phylogenetic Inference
2.3. Statistical Analysis
2.4. Rarefaction
2.5. Alpha and Beta Diversities
2.6. PERMANOVA
2.7. Differential Abundancy Analysis
3. Results
3.1. Amplicon Sequence Variants (ASVs)
3.2. Rarefaction
3.3. Community Composition
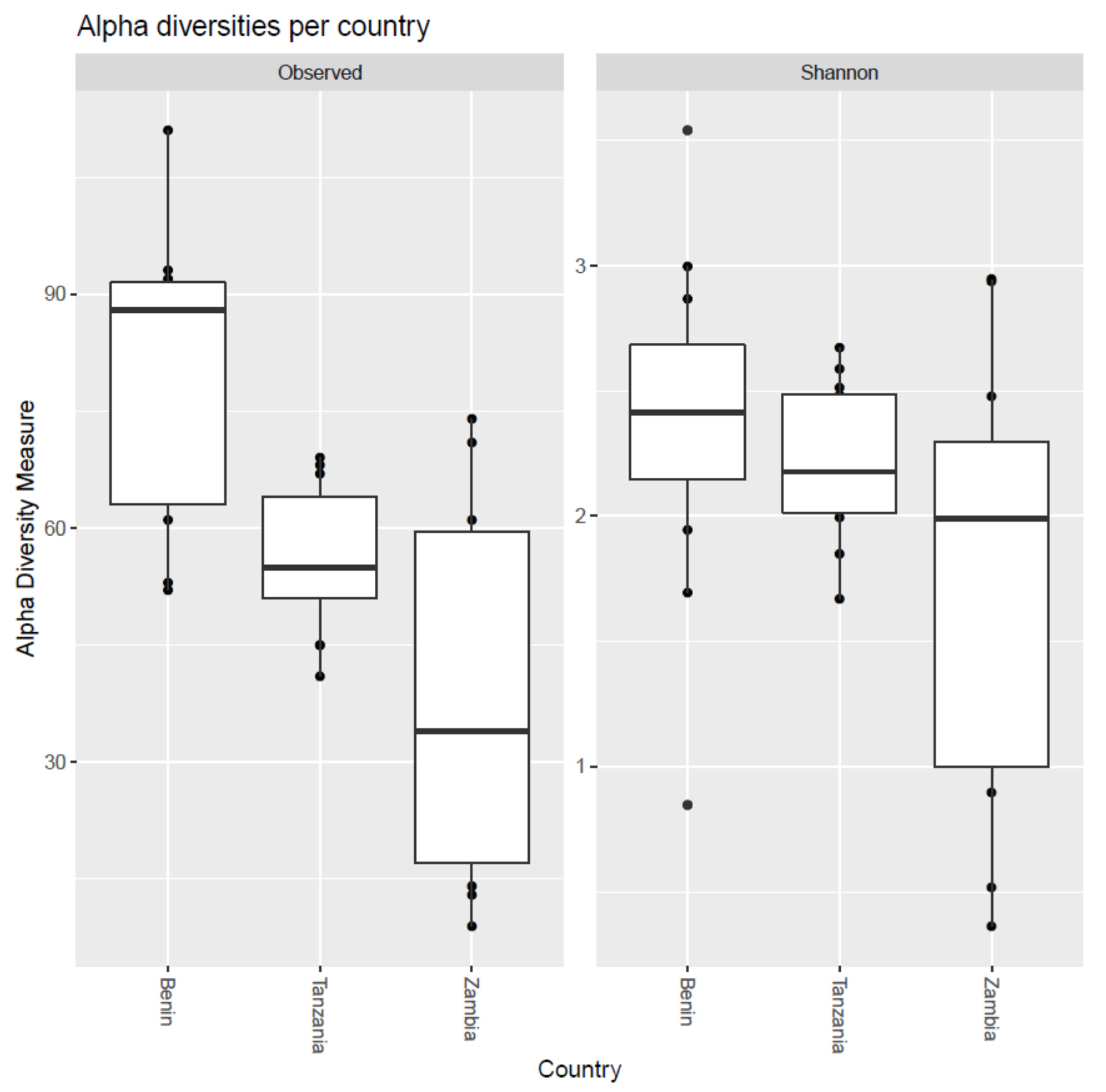
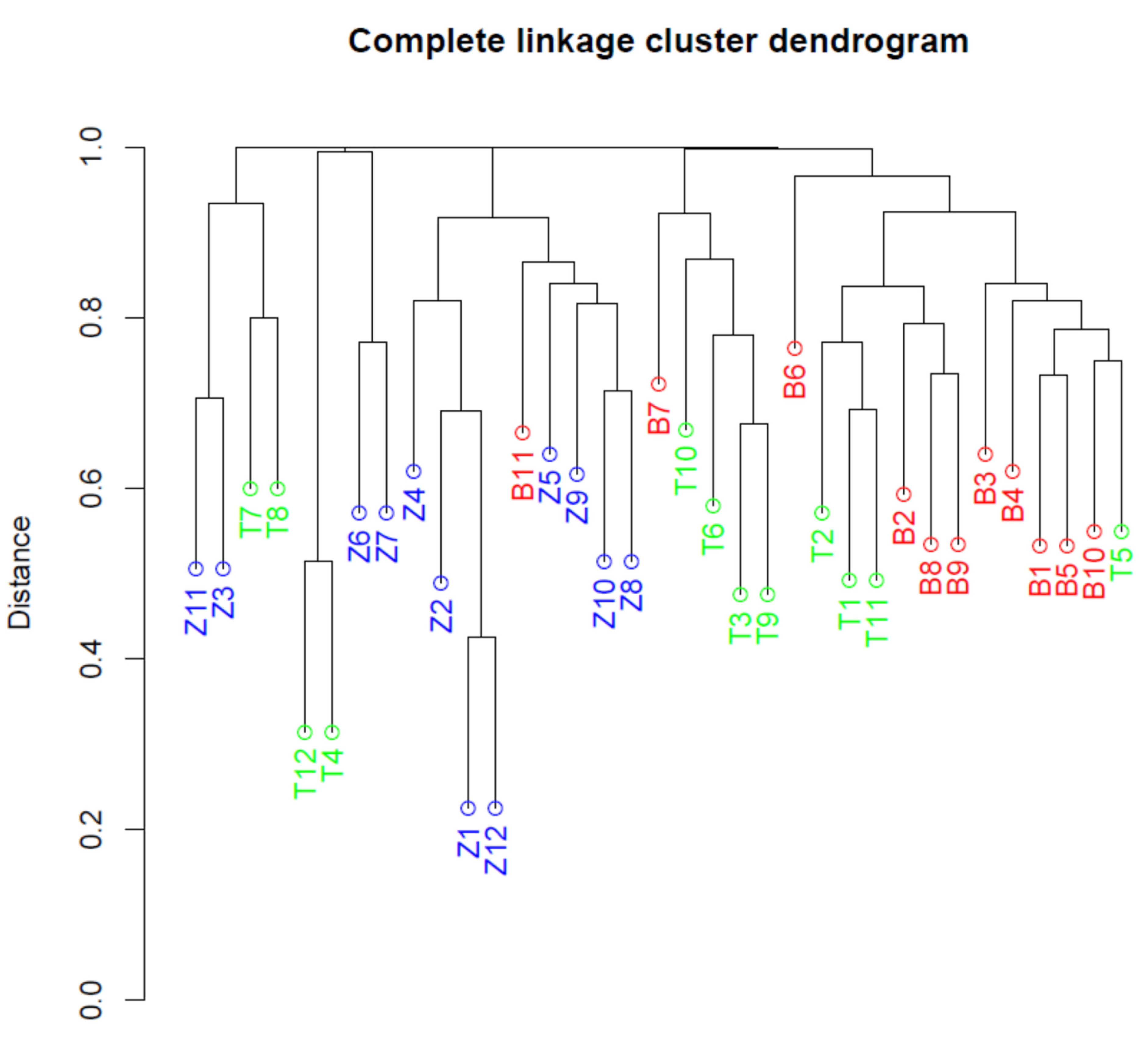
3.4. Alpha and Beta Diversity in Microbial Communities
3.5. PERMANOVA Differential Abundance Testing
3.6. Differential ASV Abundance Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolfe, B.E.; Dutton, R.J. Fermented foods as experimentally tractable microbial ecosystems. Cell 2015, 161, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alekseeva, A.Y.; Groenenboom, A.E.; Smid, E.J.; Schoustra, S.E. Eco-Evolutionary Dynamics in Microbial Communities from Spontaneous Fermented Foods. Int. J. Environ. Res. Public Health 2021, 18, 10093. [Google Scholar] [CrossRef] [PubMed]
- McGovern, P.E.; Zhang, J.; Tang, J.; Zhang, Z.; Hall, G.R.; Moreau, R.A.; Nuñ ez, A.; Butrym, E.D.; Richards, M.P.; Wang, C.; et al. Fermented beverages of pre-and proto-historic China. Proc. Natl. Acad. Sci. USA 2004, 101, 17593–17598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulová, L.; Patáková, P.; Brányik, T. Advanced Fermentation Processes. In Engineering Aspects of Food Biotechnology; Teixeira, J.A., Vicente, A.A., Eds.; Taylor & Francis Group: Boca Raton, FL, USA, 2013; pp. 85–110. [Google Scholar] [CrossRef]
- Dertli, E.; Çon, A.H. Microbial diversity of traditional kefir grains and their role on kefir aroma. LWT Food Sci. Technol. 2017, 85, 151–157. [Google Scholar] [CrossRef]
- Schoustra, S.E.; Kasase, C.; Toarta, C.; Kassen, R.; Poulain, A.J. Microbial Community Structure of Three Traditional Zambian Fermented Products: Mabisi, Chibwantu and Munkoyo. PLoS ONE 2013, 8, e63948. [Google Scholar] [CrossRef] [Green Version]
- Phiri, S.; Schoustra, S.E.; van den Heuvel, J.; Smid, E.J.; Shindano, J.; Linnemann, A. Fermented cereal-based Munkoyo beverage: Processing practices, microbial diversity and aroma compounds. PLoS ONE 2019, 14, e0223501. [Google Scholar] [CrossRef] [Green Version]
- Moonga, H.B.; Schoustra, S.E.; van den Heuvel, J.; Linnemann, A.R.; Samad, M.S.; Shindano, J.; Smid, E.J. Composition and Diversity of Natural Bacterial Communities in Mabisi, a Traditionally Fermented Milk. Front. Microbiol. 2020, 11, 1816. [Google Scholar] [CrossRef]
- Moonga, H.B.; Schoustra, S.E.; Linnemann, A.R.; van den Heuvel, J.; Shindano, J.; Smid, E.J. Influence of fermentation temperature on microbial community composition and physicochemical properties of mabisi, a traditionally fermented milk. LWT 2021, 136, 110350. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Author Correction: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Marizzoni, M.; Gurry, T.; Provasi, S.; Greub, G.; Lopizzo, N.; Ribaldi, F.; Festari, C.; Mazzelli, M.; Mombelli, E.; Salvatore, M.; et al. Comparison of Bioinformatics Pipelines and Operating Systems for the Analyses of 16S rRNA Gene Amplicon Sequences in Human Fecal Samples. Front. Microbiol. 2020, 11, 1262. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- DJ Lane 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E.; Goodfellow, M. (Eds.) John Wiley and Sons: New York, NY, USA, 1991; pp. 115–175. Available online: https://ci.nii.ac.jp/naid/10004891521/ (accessed on 10 May 2022).
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Heibl, C.; Cusimano, N.; Krah, F.-S. Package “ips” 2019. Available online: https://CRAN.R-project.org/package=ips (accessed on 10 May 2022).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Simpson, G.L.; Kindt, R.; Legendre, P.; Minchin, P. The Vegan Package. 2009. Available online: https://CRAN.R-project.org/package=vegan (accessed on 10 May 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication. Bell Syst. Tech. J. 1949, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228. [Google Scholar] [CrossRef] [Green Version]
- Prentice, I.C. Non-Metric Ordination Methods in Ecology. Source J. Ecol. 1977, 65, 85–94. [Google Scholar] [CrossRef]
- Romesburg, C. Cluster Analysis for Researchers; LULU Publishers: Morrisville, NC, 2004; ISBN 0534032486. [Google Scholar]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Noble, W.S. How does multiple testing correction work? Nat. Biotechnol. 2009, 27, 1135–1137. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gotelli, N.J.; Colwell, R.K. Quantifying biodiversity: Procedures and pitfalls in the measurement and comparison of species richness. Ecol. Lett. 2001, 4, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Dodd, C.E.R. Infrequent Microbial Infections. Foodborne Dis. Third Ed. 2017, 277–288. [Google Scholar] [CrossRef]
- Bergey, D. Bergey’s Manual of Determinative Bacteriology, 5th ed.; The Williams & Wilkins Co.: Baltimore, MD, USA, 1994; ISBN 978068341051835. [Google Scholar]
- Mounir, M.; Shafiei, R.; Zarmehrkhorshid, R.; Hamouda, A.; Alaoui, M.I.; Thonart, P. Evaluation of thermotolerant acetobacter pasteurianus strains isolated from moroccan fruits catalyzing oxidative fermentation at high temperature. Commun. Appl. Biol. Sci. 2015, 80, 37–43. [Google Scholar] [PubMed]
- Khan, I.U.; Habib, N.; Asem, M.D.; Salam, N.; Xiao, M.; Zhou, E.M.; Zhi, X.Y.; Li, W.J. Aquabacterium tepidiphilum sp. nov., a moderately thermophilic bacterium isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2019, 69, 337–342. [Google Scholar] [CrossRef]
- Shah, A.A.; Nawaz, A.; Kanwal, L.; Hasan, F.; Khan, S.; Badshah, M. Degradation of poly(ε-caprolactone) by a thermophilic bacterium Ralstonia sp. strain MRL-TL isolated from hot spring. Int. Biodeterior. Biodegrad. 2015, 98, 35–42. [Google Scholar] [CrossRef]
- Groenenboom, A.E.; Parker, M.E.; de Vries, A.; de Groot, S.; Zobrist, S.; Mansen, K.; Milani, P.; Kort, R.; Smid, E.J.; Schoustra, S.E. Bacterial community dynamics in lait caillé, a traditional product of spontaneous fermentation from Senegal. PLoS ONE 2019, 14, e0215658. [Google Scholar] [CrossRef] [PubMed]
- Moonga, H.B.; Phiri, S.; Schoustra, S.E.; Chileshe, J.; Chirwa-Moonga, T.; Shindano, J. The Munkoyo Root: Traditional Uses, Biochemistry, Fermentation, and Potential Cultivation. ACS Symp. Ser. 2020, 1361, 81–99. [Google Scholar] [CrossRef]
- Freilich, S.; Kreimer, A.; Meilijson, I.; Gophna, U.; Sharan, R.; Ruppin, E. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res. 2010, 38, 3857–3868. [Google Scholar] [CrossRef] [PubMed]
- Elith, J.; Leathwick, J.R. Species Distribution Models: Ecological Explanation and Prediction Across Space and Time. Curr. Sci. 2009, 40, 677–697. [Google Scholar] [CrossRef]
- Groenenboom, A.E.; Shindano, J.; Cheepa, N.; Smid, E.J.; Schoustra, S.E. Microbial population dynamics during traditional production of Mabisi, a spontaneous fermented milk product from Zambia: A field trial. World J. Microbiol. Biotechnol. 2020, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Materia, V.C.; Linnemann, A.R.; Smid, E.J.; Schoustra, S.E. Contribution of traditional fermented foods to food systems transformation: Value addition and inclusive entrepreneurship. Food Secur. 2021, 13, 1163–1177. [Google Scholar] [CrossRef]
- Nout, M.J.R.; Sarkar, P.K.; Beuchat, L.R. Indigenous fermented foods food microbiology. In Food Microbiology: Fundamentals and Frontiers; Doyle, M.P., Beuchat, L.R., Eds.; ASM Press: Washingon, DC, USA, 2007; pp. 817–835. [Google Scholar]
- Jones, E.W.; Carlson, J.M.; Sivak, D.A.; Ludington, W.B. Stochastic microbiome assembly depends on context. Proc. Natl. Acad. Sci. USA 2022, 119, e2115877119. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Minimum | First Quartile | Median | Third Quartile | Mean | Maximum |
|---|---|---|---|---|---|
| 1 | 12 | 34 | 113 | ~530 | 73,787 |
| # Amplicons Total | # Amplicons in Samples | Number of ASVs | |
|---|---|---|---|
| Before rarefaction | 597,755 | 5021–44,710 | 1127 |
| After rarefaction | 158,130 | 4518 | 1070 |
| Minimum | First Quantile | Median | Third Median | Mean | Maximum |
|---|---|---|---|---|---|
| 1 | 4 | 20 | 157 | ~838 | 35,696 |
| All Countries | Benin vs. Tanzania | Benin vs. Zambia | Tanzania vs. Zambia | |
|---|---|---|---|---|
| Between group variation | 0.185 | 0.072 | 0.179 | 0.183 |
| Within group variation | 0.815 | 0.928 | 0.821 | 0.817 |
| Total variation | 1 | 1 | 1 | 1 |
| F value | 3.628 | 1.634 | 4.582 | 4.933 |
| Pr (>F) | 0.001 | 0.015 | 0.001 | 0.001 |
| Country A | Country B | Overabundant ASVs in A | Overabundant ASVs in B | Total Differentially Abundant ASVs | Total Number of ASVs |
|---|---|---|---|---|---|
| Benin | Tanzania | 30 | 22 | 52 | 858 |
| Benin | Zambia | 60 | 31 | 91 | 803 |
| Tanzania | Zambia | 53 | 27 | 80 | 654 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Jong, M.; Alekseeva, A.Y.; Miraji, K.F.; Phiri, S.; Linnemann, A.R.; Schoustra, S.E. Environmental Selection Shapes Bacterial Community Composition in Traditionally Fermented Maize-Based Foods from Benin, Tanzania and Zambia. Microorganisms 2022, 10, 1354. https://doi.org/10.3390/microorganisms10071354
de Jong M, Alekseeva AY, Miraji KF, Phiri S, Linnemann AR, Schoustra SE. Environmental Selection Shapes Bacterial Community Composition in Traditionally Fermented Maize-Based Foods from Benin, Tanzania and Zambia. Microorganisms. 2022; 10(7):1354. https://doi.org/10.3390/microorganisms10071354
Chicago/Turabian Stylede Jong, Maarten, Anna Y. Alekseeva, Kulwa F. Miraji, Sydney Phiri, Anita R. Linnemann, and Sijmen E. Schoustra. 2022. "Environmental Selection Shapes Bacterial Community Composition in Traditionally Fermented Maize-Based Foods from Benin, Tanzania and Zambia" Microorganisms 10, no. 7: 1354. https://doi.org/10.3390/microorganisms10071354