
Population Structure and Genomic Characteristics of Australian Erysipelothrix rhusiopathiae Reveals Unobserved Diversity in the Australian Pig Industry
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibiotic Resistance Profiling
2.2. Extraction and Purification of DNA for WGS
2.3. Genome Assembly and Annotation
2.4. Identification of SpaA, Surface Proteins and Antimicrobial Resistance Genes
2.5. Multi Locus Sequence Typing
2.6. Phylogenetic Inference
2.7. Estimating Recombination Rates
3. Results
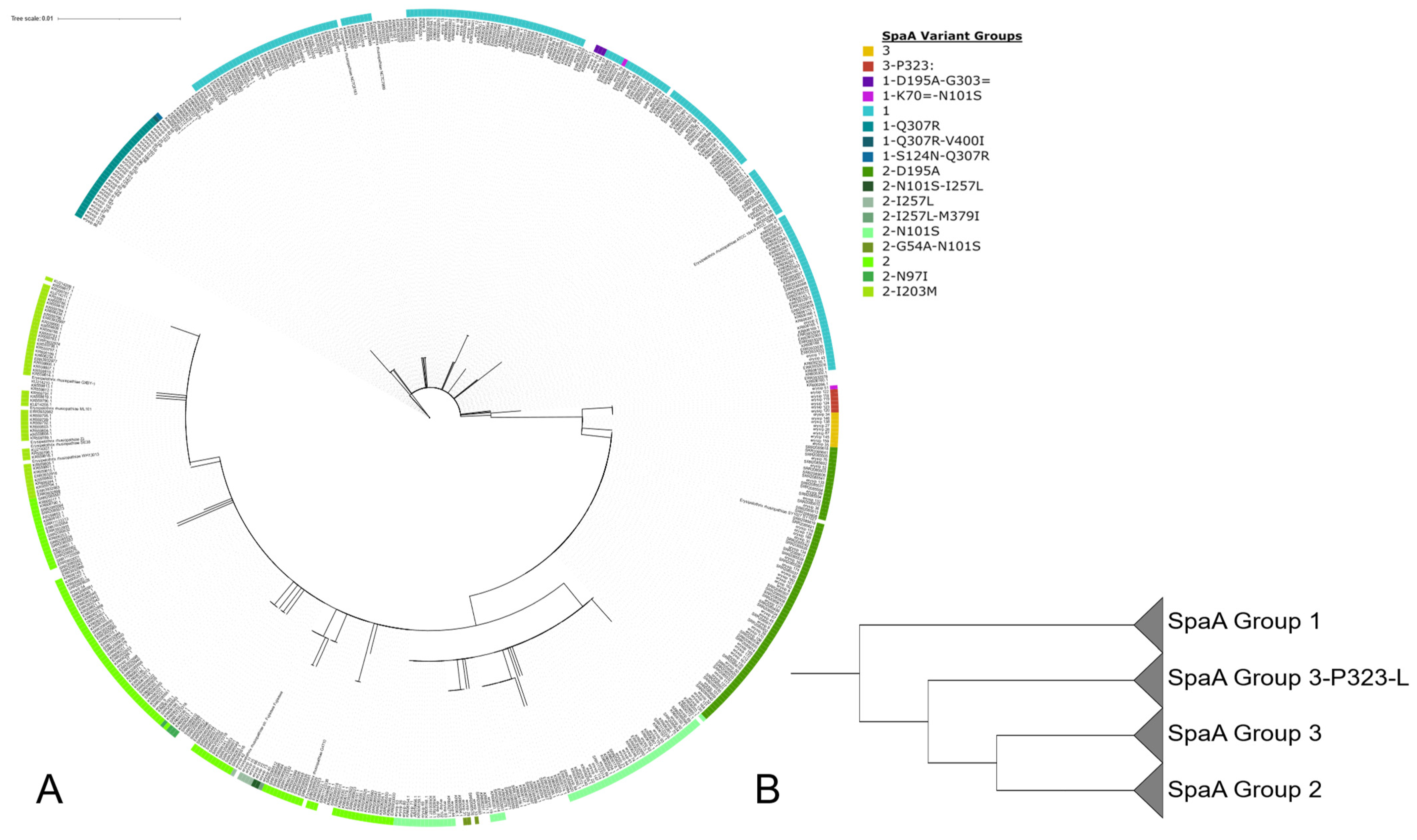
3.1. Population Structure of E. rhusiopathiae
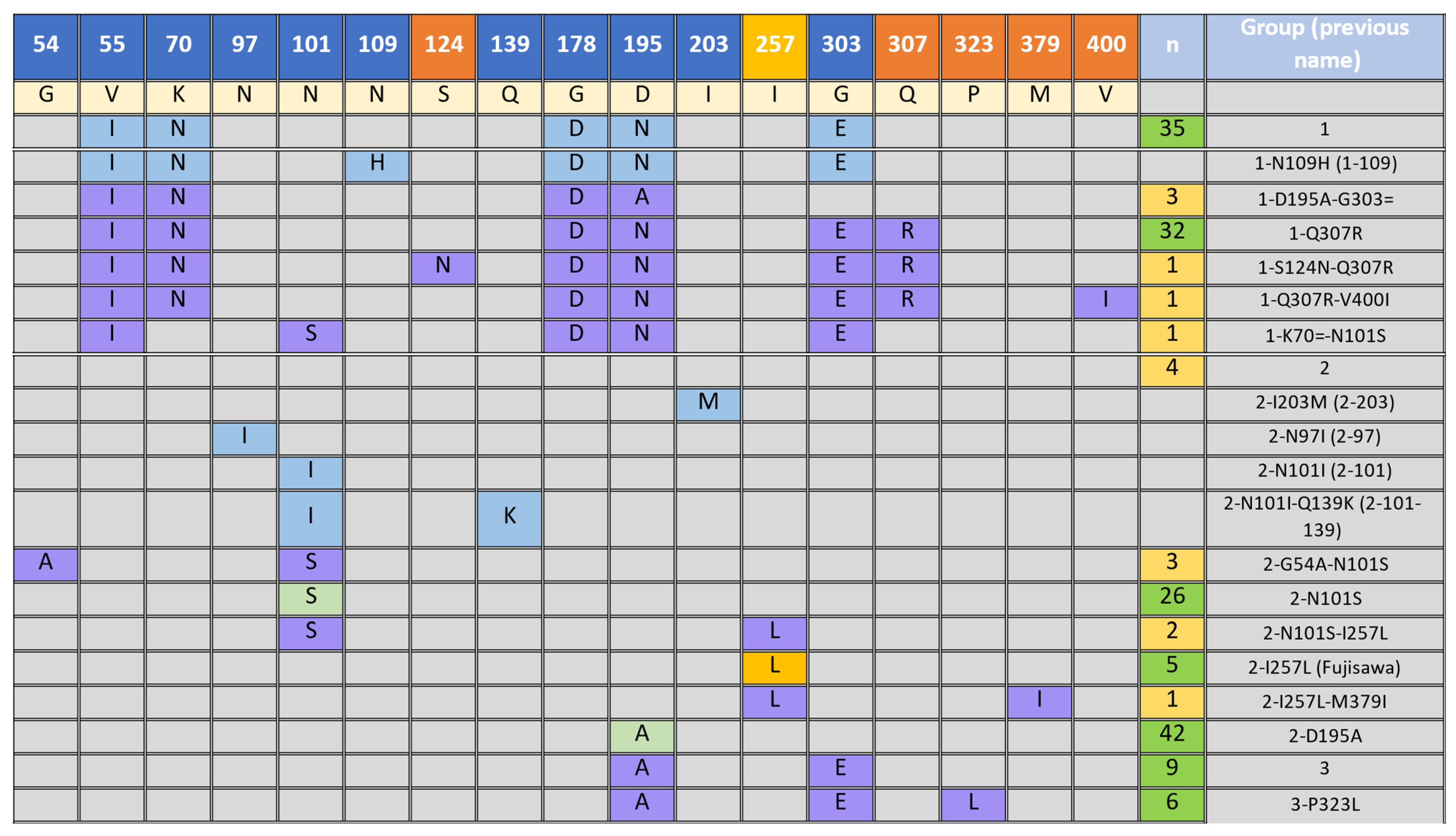
3.2. Presence of SpaA Proteins in E. rhusiopathiae
3.3. Recombination Rates in E. rhusipathiae Clade 2, 3 and Australian Clades
3.4. Vaccine Breakdown amongst Spa Groups
3.5. Surface Protein Screening
3.6. Resistance Gene Screening
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Forde, T.L.; Orsel, K.; Zadoks, R.N.; Biek, R.; Adams, L.G.; Checkley, S.L.; Davison, T.; De Buck, J.; Dumond, M.; Elkin, B.T.; et al. Bacterial Genomics Reveal the Complex Epidemiology of an Emerging Pathogen in Arctic and Boreal Ungulates. Front. Microbiol. 2016, 7, 1759. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Jansson, D.S.; Johansson, K.-E.; Båverud, V.; Chirico, J.; Aspán, A. Characterization of Erysipelothrix rhusiopathiae isolates from poultry, pigs, emus, the poultry red mite and other animals. Vet. Microbiol. 2009, 137, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Rosenbach, F. Experimentelle, morphologische und klinische Studie über die krankheitserregenden Mikroorganismen des Schweinerotlaufs, des Erysipeloids und der Mäusesepsis. Z. Hyg. Infekt. 1909, 63, 343–371. [Google Scholar] [CrossRef]
- Forde, T.L.; Kollanandi Ratheesh, N.; Harvey, W.T.; Thomson, J.R.; Williamson, S.; Biek, R.; Opriessnig, T. Genomic and immunogenic protein diversity of Erysipelothrix rhusiopathiae isolated from pigs in Great Britain: Implications for vaccine protection. Front. Microbiol. 2020, 11, 418. [Google Scholar] [CrossRef]
- Ogawa, Y.; Ooka, T.; Shi, F.; Ogura, Y.; Nakayama, K.; Hayashi, T.; Shimoji, Y. The genome of Erysipelothrix rhusiopathiae, the causative agent of swine erysipelas, reveals new insights into the evolution of Firmicutes and the organism’s intracellular adaptations. J. Bacteriol. 2011, 193, 2959–2971. [Google Scholar] [CrossRef]
- Opriessnig, T.; Forde, T.; Shimoji, Y. Erysipelothrix spp.: Past, present, and future directions in vaccine research. Front. Vet. Sci. 2020, 7, 174. [Google Scholar] [CrossRef]
- Yang, L.; Zhu, Y.; Peng, Z.; Ding, Y.; Jie, K.; Wang, Z.; Peng, Y.; Tang, X.; Wang, X.; Chen, H.; et al. Comparative Genome Analysis of a Pathogenic Erysipelothrix rhusiopathiae Isolate WH13013 from Pig Reveals Potential Genes Involve in Bacterial Adaptions and Pathogenesis. Vet. Sci. 2020, 7, 74. [Google Scholar] [CrossRef]
- Kwok, A.H.; Li, Y.; Jiang, J.; Jiang, P.; Leung, F.C. Complete genome assembly and characterization of an outbreak strain of the causative agent of swine erysipelas–Erysipelothrix rhusiopathiae SY1027. BMC Microbiol. 2014, 14, 176. [Google Scholar] [CrossRef]
- Ogawa, Y.; Shiraiwa, K.; Ogura, Y.; Ooka, T.; Nishikawa, S.; Eguchi, M.; Hayashi, T.; Shimoji, Y. Clonal lineages of Erysipelothrix rhusiopathiae responsible for acute swine erysipelas in Japan identified by using genome-wide single-nucleotide polymorphism analysis. Appl. Environ. Microbiol. 2017, 83, e00130-17. [Google Scholar] [CrossRef]
- Bender, J.; Shen, H.; Irwin, C.; Schwartz, K.; Opriessnig, T. Characterization of Erysipelothrix species isolates from clinically affected pigs, environmental samples, and vaccine strains from six recent swine erysipelas outbreaks in the United States. Clin. Vaccine Immunol. 2010, 17, 1605–1611. [Google Scholar] [CrossRef]
- Eriksson, H.; Brännström, S.; Skarin, H.; Chirico, J. Characterization of Erysipelothrix rhusiopathiae isolates from laying hens and poultry red mites (Dermanyssus gallinae) from an outbreak of erysipelas. Avian Pathol. 2010, 39, 505–509. [Google Scholar] [CrossRef]
- Gerber, P.F.; MacLeod, A.; Opriessnig, T. Erysipelothrix rhusiopathiae serotype 15 associated with recurring pig erysipelas outbreaks. Vet. Rec. 2018, 182, 635. [Google Scholar] [CrossRef]
- Kutz, S.; Bollinger, T.; Branigan, M.; Checkley, S.; Davison, T.; Dumond, M.; Elkin, B.; Forde, T.; Hutchins, W.; Niptanatiak, A.; et al. Erysipelothrix rhusiopathiae associated with recent widespread muskox mortalities in the Canadian Arctic. Can. Vet. J. 2015, 56, 560. [Google Scholar]
- Langford, E.; Dorward, W. Erysipelothrix insidiosa recovered from sylvatic mammals in northwestern Canada during examinations for rabies and anthrax. Can. Vet. J. 1977, 18, 101. [Google Scholar]
- Hoffmann, C.; Bilkei, G. Case study: Chronic erysipelas of the sow–a subclinical manifestation of reproductive problems. Reprod. Domest. Anim. 2002, 37, 119–120. [Google Scholar] [CrossRef]
- Eamens, G.; Forbes, W.; Djordjevic, S. Characterisation of Erysipelothrix rhusiopathiae isolates from pigs associated with vaccine breakdowns. Vet. Microbiol. 2006, 115, 329–338. [Google Scholar] [CrossRef]
- Sawada, T.; Takahashi, T. Cross protection of mice and swine inoculated with culture filtrate of attenuated Erysipelothrix rhusiopathiae and challenge exposed to strains of various serovars. Am. J. Vet. Res. 1987, 48, 239–242. [Google Scholar]
- Shimoji, Y.; Mori, Y.; Fischetti, V.A. Immunological characterization of a protective antigen of Erysipelothrix rhusiopathiae: Identification of the region responsible for protective immunity. Infection 1999, 67, 1646–1651. [Google Scholar] [CrossRef]
- Makino, S.-i.; Yamamoto, K.; Murakami, S.; Shirahata, T.; Uemura, K.; Sawada, T.; Wakamoto, H.; Morita, Y. Properties of repeat domain found in a novel protective antigen, SpaA, of Erysipelothrix rhusiopathiae. Microb. Pathog. 1998, 25, 101–109. [Google Scholar] [CrossRef]
- Janßen, T.; Voss, M.; Kühl, M.; Semmler, T.; Philipp, H.-C.; Ewers, C. A combinational approach of multilocus sequence typing and other molecular typing methods in unravelling the epidemiology of Erysipelothrix rhusiopathiae strains from poultry and mammals. Vet. Res. 2015, 46, 84. [Google Scholar] [CrossRef]
- To, H.; Sato, H.; Tazumi, A.; Tsutsumi, N.; Nagai, S.; Iwata, A.; Nagano, T. Characterization of Erysipelothrix rhusiopathiae strains isolated from recent swine erysipelas outbreaks in Japan. J. Vet. Med. Sci. 2012, 74, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Ingebritson, A.L.; Roth, J.A.; Hauer, P.J. Erysipelothrix rhusiopathiae: Association of Spa-type with serotype and role in protective immunity. Vaccine 2010, 28, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, S.N.; Babii, A.V. Draft genome sequence data and comparative analysis of Erysipelothrix rhusiopathiae vaccine strain VR-2. 3 Biotech 2020, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Gatus, B.J.; Pham, J.N.; Rafferty, D.L. Antibiotic Susceptibility Testing by the CDS Method: A Concise Laboratory Manual; Arthur Productions Pty Ltd.: Sydney, Australia; South Eastern Area Laboratory Services: Randwick, Australia, 1999. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of genome assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Hunt, M.; Mather, A.E.; Sánchez-Busó, L.; Page, A.J.; Parkhill, J.; Keane, J.A.; Harris, S.R. ARIBA: Rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genom. 2017, 3, e000131. [Google Scholar] [CrossRef]
- Bushnell, B. BBTools Software Package. 2014. Available online: https://sourceforge.net/projects/bbmap (accessed on 12 January 2022).
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Seeman, T. ABRicate. 2020. Available online: https://github.com/tseemann/abricate (accessed on 6 April 2021).
- Seeman, T. mlst. 2020. Available online: https://github.com/tseemann/mlst (accessed on 24 February 2020).
- Ribeiro-Gonçalves, B.; Francisco Alexandre, P.; Vaz, C.; Ramirez, M.; Carriço, J.A. PHYLOViZ Online: Web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 2016, 44, W246–W251. [Google Scholar] [CrossRef]
- Webster, J.; Bogema, D. Core_gene_phylo, v1.0.0. Github. 2022. Available online: https://github.com/Jwebster89/Core_gene_phylo (accessed on 23 February 2022).
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Wilson, D.J. ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 2015, 11, e1004041. [Google Scholar] [CrossRef]
- Mai, U.; Sayyari, E.; Mirarab, S. Minimum variance rooting of phylogenetic trees and implications for species tree reconstruction. PLoS ONE 2017, 12, e0182238. [Google Scholar] [CrossRef]
- Lin, M.; Kussell, E. Inferring bacterial recombination rates from large-scale sequencing datasets. Nat. Methods 2019, 16, 199–204. [Google Scholar] [CrossRef]
- Uchiyama, M.; Shimazaki, Y.; Isshiki, Y.; Kojima, A.; Hirano, F.; Yamamoto, K.; Kijima, M.; Nagai, H. Pathogenic characterization of Erysipelothrix rhusiopathiae Met-203 type SpaA strains from chronic and subacute swine erysipelas in Japan. J. Vet. Med. Sci. 2017, 79, 18–21. [Google Scholar] [CrossRef]
- To, H.; Nagai, S. Genetic and antigenic diversity of the surface protective antigen proteins of Erysipelothrix rhusiopathiae. Clin. Vaccine Immunol. 2007, 14, 813–820. [Google Scholar] [CrossRef]
- Jeon, W.; Kim, Y.-C.; Hong, M.; Rejinold, S.; Park, K.; Yoon, I.; Yoo, S.; Lee, H.; Ahn, J. Microcrystalline cellulose for delivery of recombinant protein-based antigen against erysipelas in mice. BioMed Res. Int. 2018, 2018, 7670505. [Google Scholar] [CrossRef]
- Shi, F.; Ogawa, Y.; Sano, A.; Harada, T.; Hirota, J.; Eguchi, M.; Oishi, E.; Shimoji, Y. Characterization and identification of a novel candidate vaccine protein through systematic analysis of extracellular proteins of Erysipelothrix rhusiopathiae. Infect. Immun. 2013, 81, 4333–4340. [Google Scholar] [CrossRef]
- Söderlund, R.; Formenti, N.; Caló, S.; Chiari, M.; Zoric, M.; Alborali, G.L.; Dalgaard, T.S.; Wattrang, E.; Eriksson, H. Comparative genome analysis of Erysipelothrix rhusiopathiae isolated from domestic pigs and wild boars suggests host adaptation and selective pressure from the use of antibiotics. Microb. Genom. 2020, 6, mgen000412. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Vidal, N.M.; Hoepers, P.G.; Reis, T.F.; Mesa, D.; Caron, L.F.; Ingberman, M.; Beirão, B.C.; Zuffo, J.P.; Fonseca, B.B. Comparative genomics of a novel clade shed light on the evolution of the genus Erysipelothrix and characterise an emerging species. Sci. Rep. 2021, 11, 3383. [Google Scholar] [CrossRef]
- Smith, J.T.; Eckhardt, E.M.; Hansel, N.B.; Eliato, T.R.; Martin, I.W.; Andam, C.P. Genomic epidemiology of methicillin-resistant and-susceptible Staphylococcus aureus from bloodstream infections. BMC Infect. Dis. 2021, 21, 589. [Google Scholar] [CrossRef]
- Park, C.J.; Andam, C.P. Distinct but intertwined evolutionary histories of multiple Salmonella enterica subspecies. Msystems 2020, 5, e00515-19. [Google Scholar] [CrossRef]
- Turner, A. Quarantine, exports and animal disease in Australia 1901–2010. Aust. Vet. J. 2011, 89, 366–371. [Google Scholar] [CrossRef]
- Takahashi, T.; Sawada, T.; Ohmae, K.; Terakado, N.; Muramatsu, M.; Seto, K.; Maruyama, T.; Kanzaki, M. Antibiotic resistance of Erysipelothrix rhusiopathiae isolated from pigs with chronic swine erysipelas. Antimicrob. Agents Chemother. 1984, 25, 385–386. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Vaccine Breakdown | Total | Breakdown (%) | Herds * | Breakdown Herd * | Breakdown Herd * (%) | Reference |
|---|---|---|---|---|---|---|---|
| 1 ** | 7 | 35 | 17.14 | 25 | 5 | 20 | Forde et al., 2020 [4] |
| 2 ** | 1 | 4 | 25 | 4 | 1 | 25 | Forde et al., 2020 [4] |
| 3 | 5 | 9 | 55.56 | 7 | 3 | 42.86 | This study |
| 1-S124N-Q307R | 1 | 1 | 100 | 1 | 1 | 100 | This study |
| 1-D195A-G303= | 1 | 3 | 33.33 | 3 | 1 | 33.33 | This study |
| 1-Q307R | 8 | 32 | 25 | 17 | 6 | 35.30 | This study |
| 1-Q307R-V400I | 1 | 1 | 100 | 1 | 1 | 100 | This study |
| 1-K70 = -N101S | 0 | 1 | 0 | 1 | 0 | 0 | This study |
| 2-N101S ** | 14 | 26 | 53.85 | 19 | 10 | 52.63 | Janßen et al., 2015 [20] |
| 2-N101S-I257L | 2 | 2 | 100 | 1 | 1 | 100 | This study |
| 2-D195A ** | 3 | 42 | 7.14 | 20 | 3 | 15 | Uchiyama et al., 2016 [41] |
| 2-I257L ** | 0 | 5 | 0 | 4 | 0 | 0 | Fujisawa reference sequence |
| 2-I257L-M379I | 0 | 1 | 0 | 1 | 0 | 0 | This study |
| 2-G54A-N101S | 0 | 3 | 0 | 3 | 0 | 0 | This study |
| 3-P323L | 6 | 6 | 100 | 1 | 1 | 100 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webster, J.; Bowring, B.; Stroud, L.; Marsh, I.; Sales, N.; Bogema, D. Population Structure and Genomic Characteristics of Australian Erysipelothrix rhusiopathiae Reveals Unobserved Diversity in the Australian Pig Industry. Microorganisms 2023, 11, 297. https://doi.org/10.3390/microorganisms11020297
Webster J, Bowring B, Stroud L, Marsh I, Sales N, Bogema D. Population Structure and Genomic Characteristics of Australian Erysipelothrix rhusiopathiae Reveals Unobserved Diversity in the Australian Pig Industry. Microorganisms. 2023; 11(2):297. https://doi.org/10.3390/microorganisms11020297
Chicago/Turabian StyleWebster, John, Bethany Bowring, Leah Stroud, Ian Marsh, Narelle Sales, and Daniel Bogema. 2023. "Population Structure and Genomic Characteristics of Australian Erysipelothrix rhusiopathiae Reveals Unobserved Diversity in the Australian Pig Industry" Microorganisms 11, no. 2: 297. https://doi.org/10.3390/microorganisms11020297