

Combined Analysis of the Whole Transcriptome of Piglets Infected with SADS−CoV Virulent and Avirulent Strains
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Strain
2.2. Piglet Challenge Experiments
2.3. Library Construction and Sequencing Process
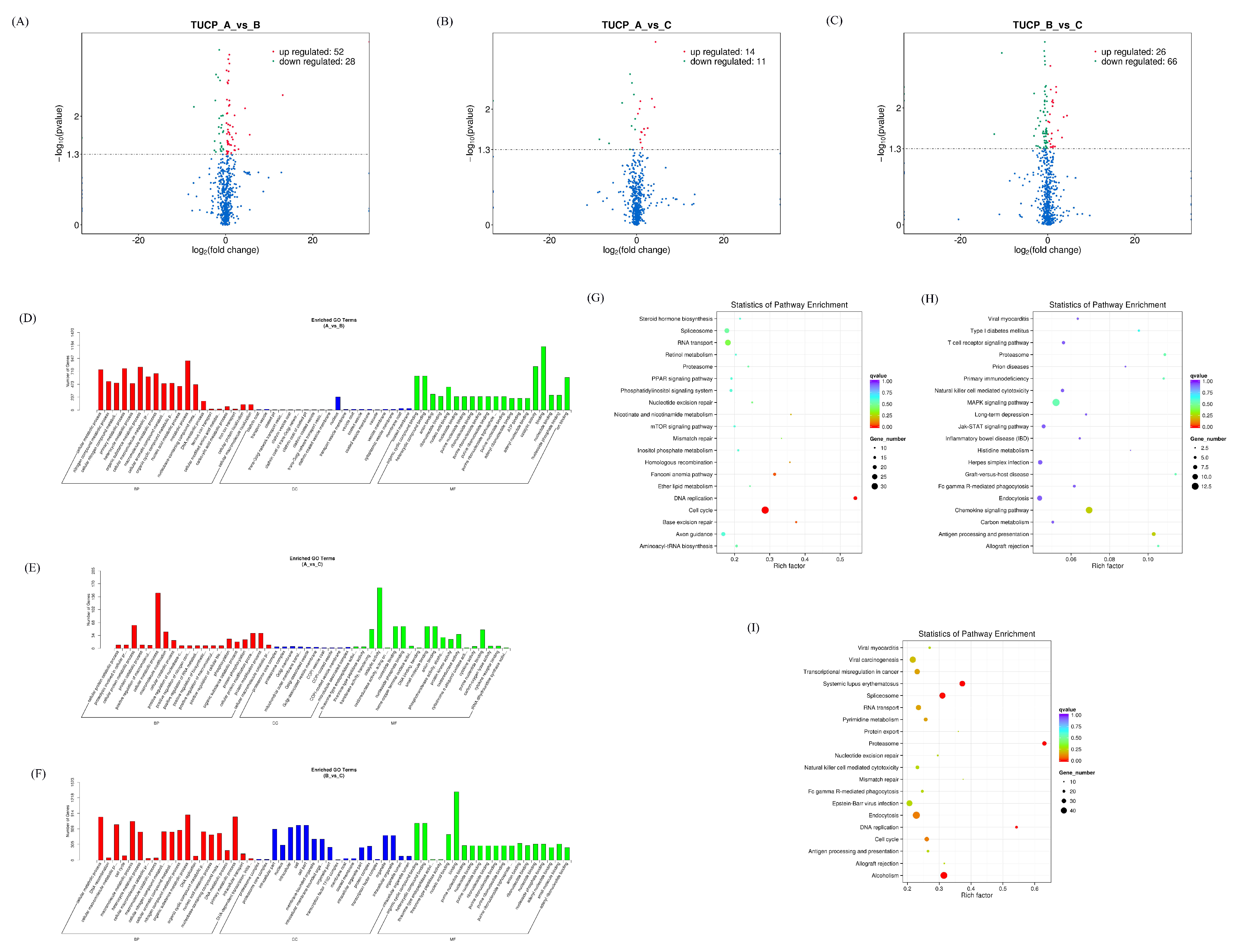
2.4. Differentially Expressed RNA Screening
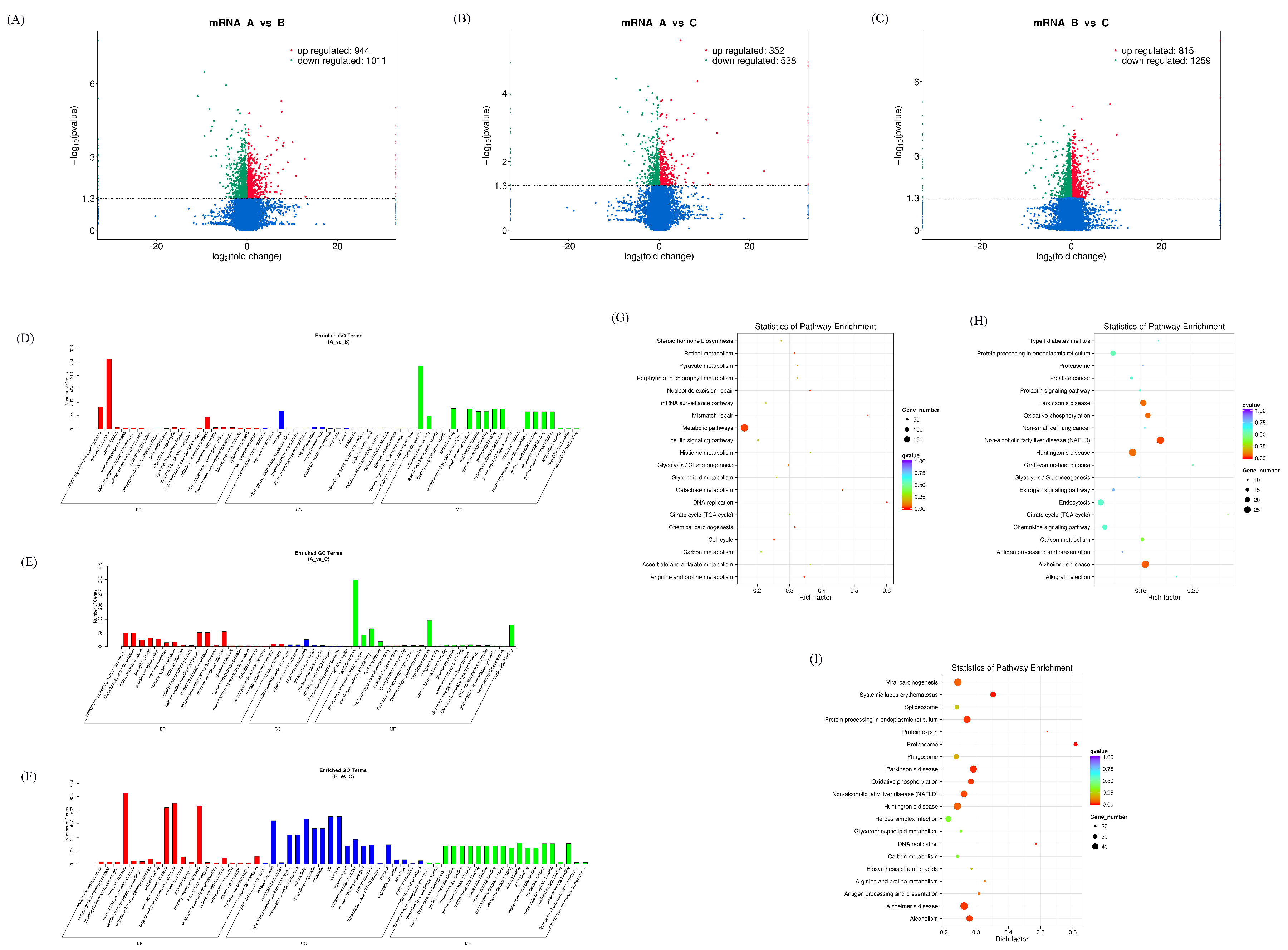
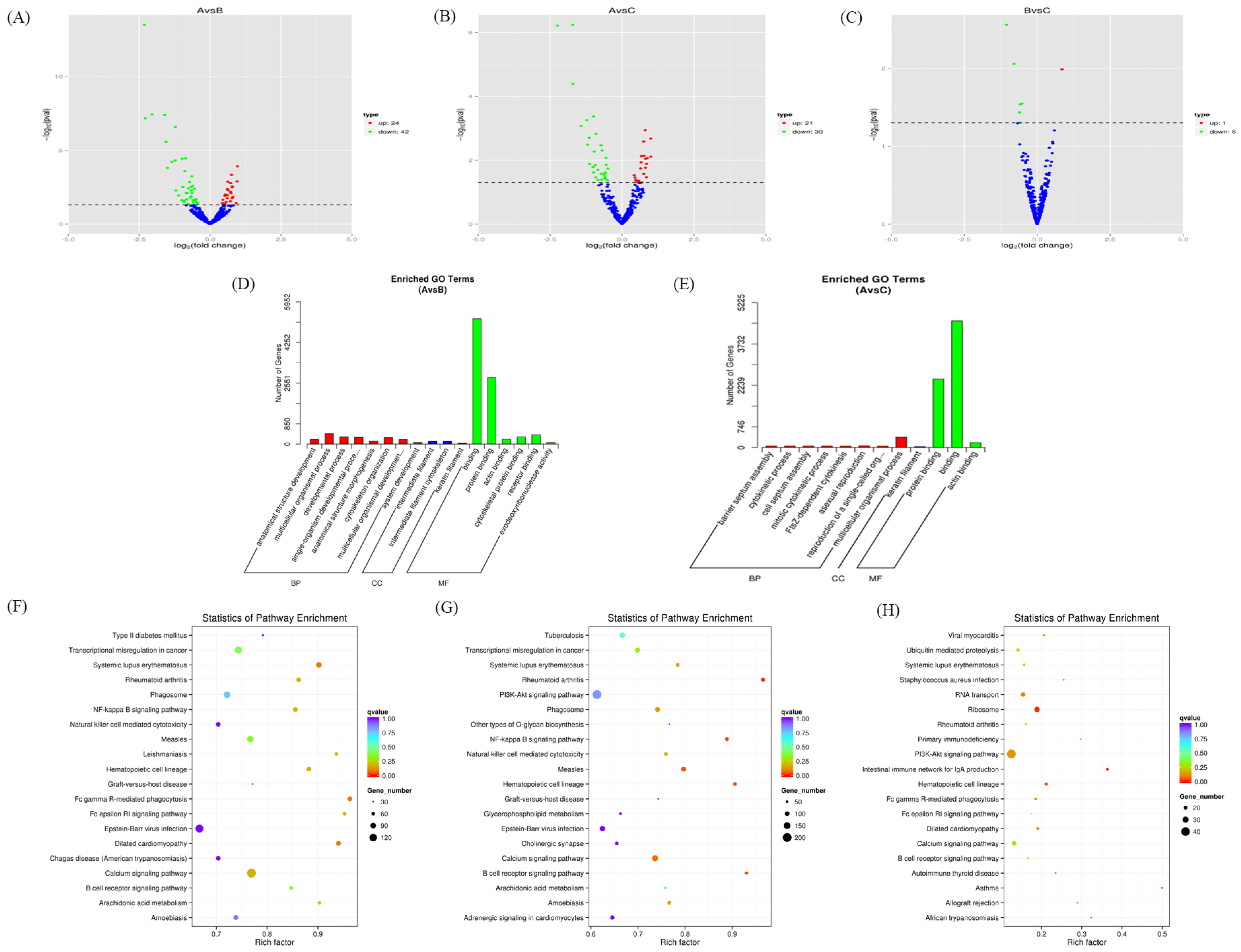
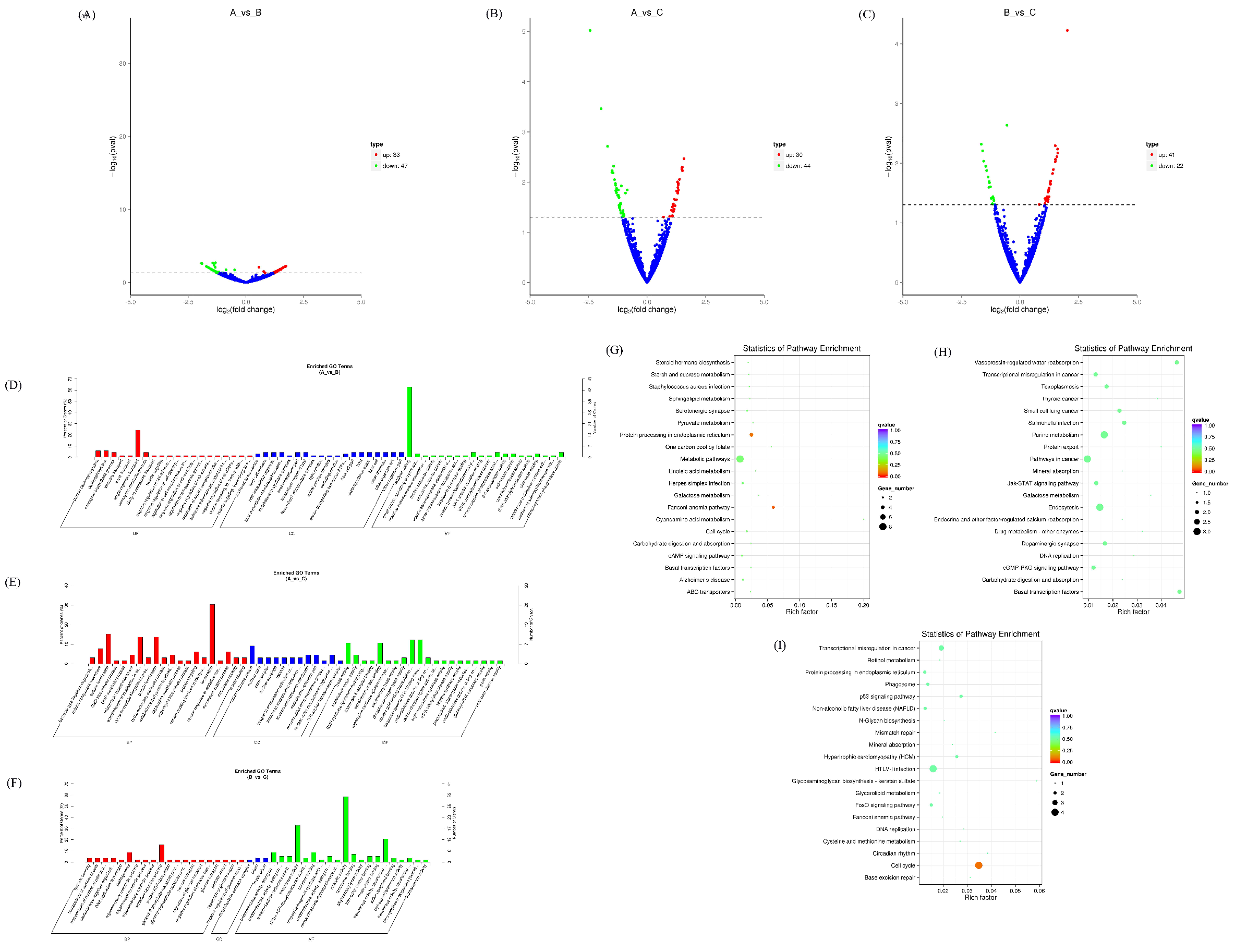
2.5. GO and KEGG Enrichment Analysis
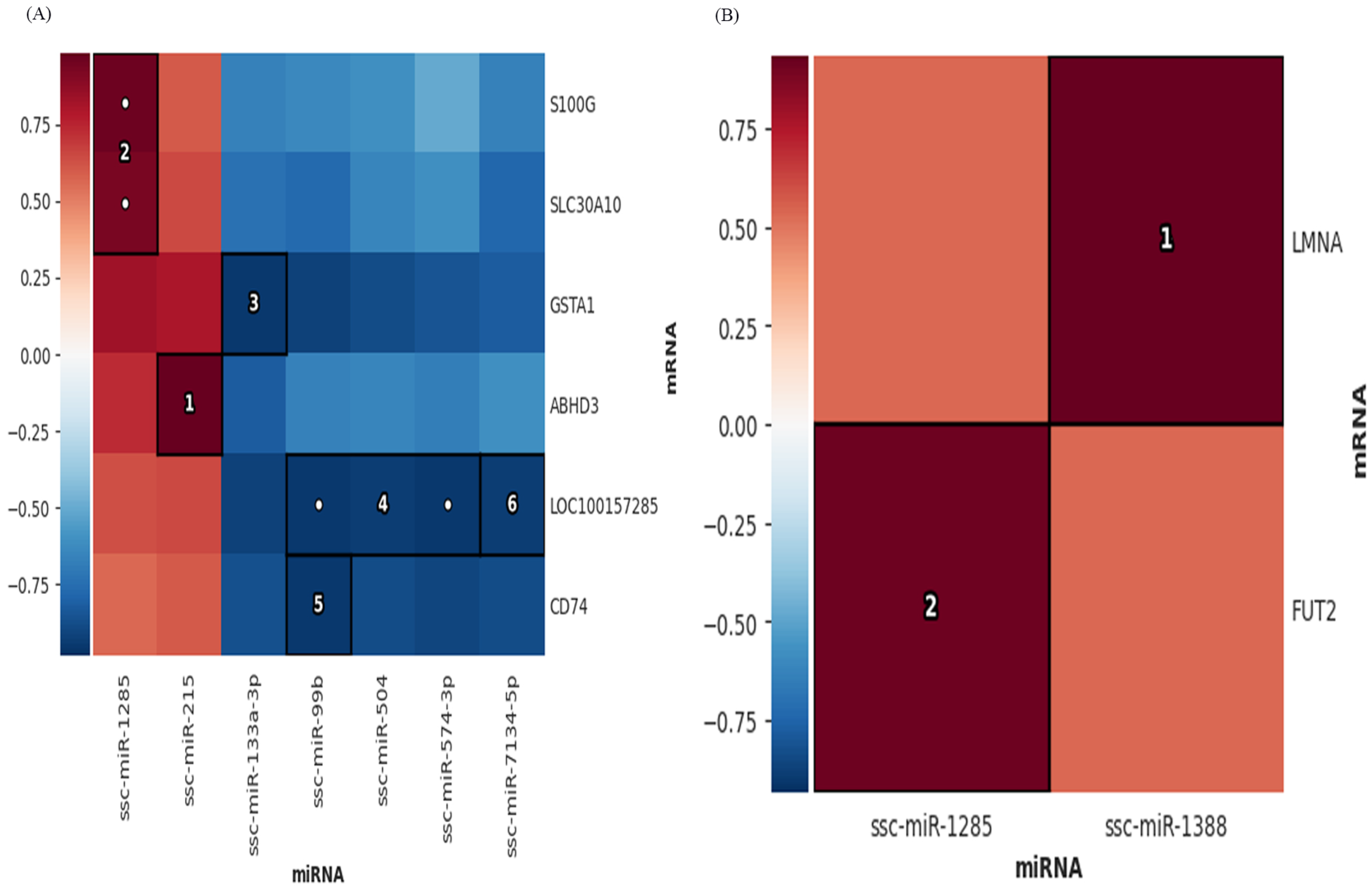
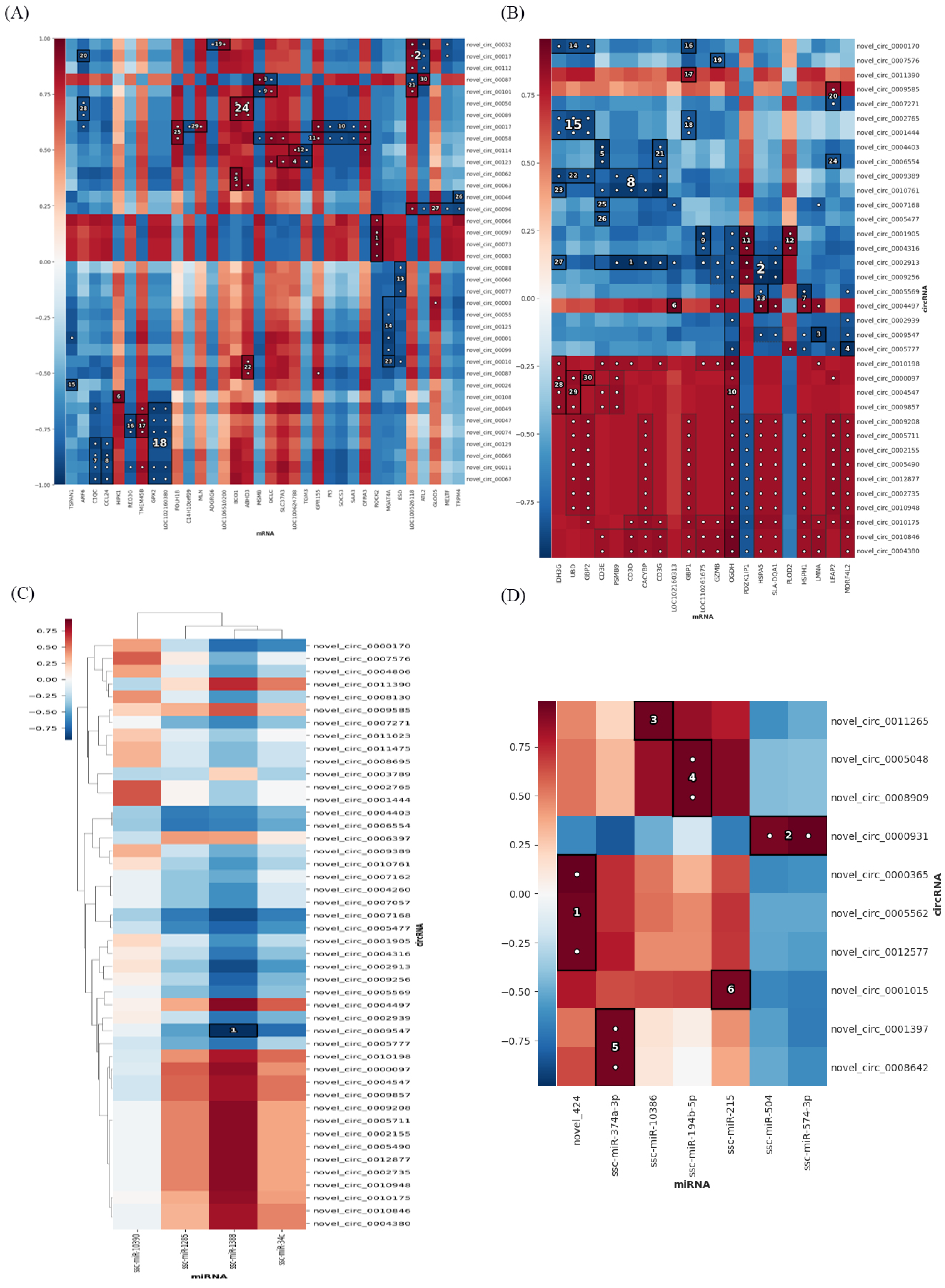
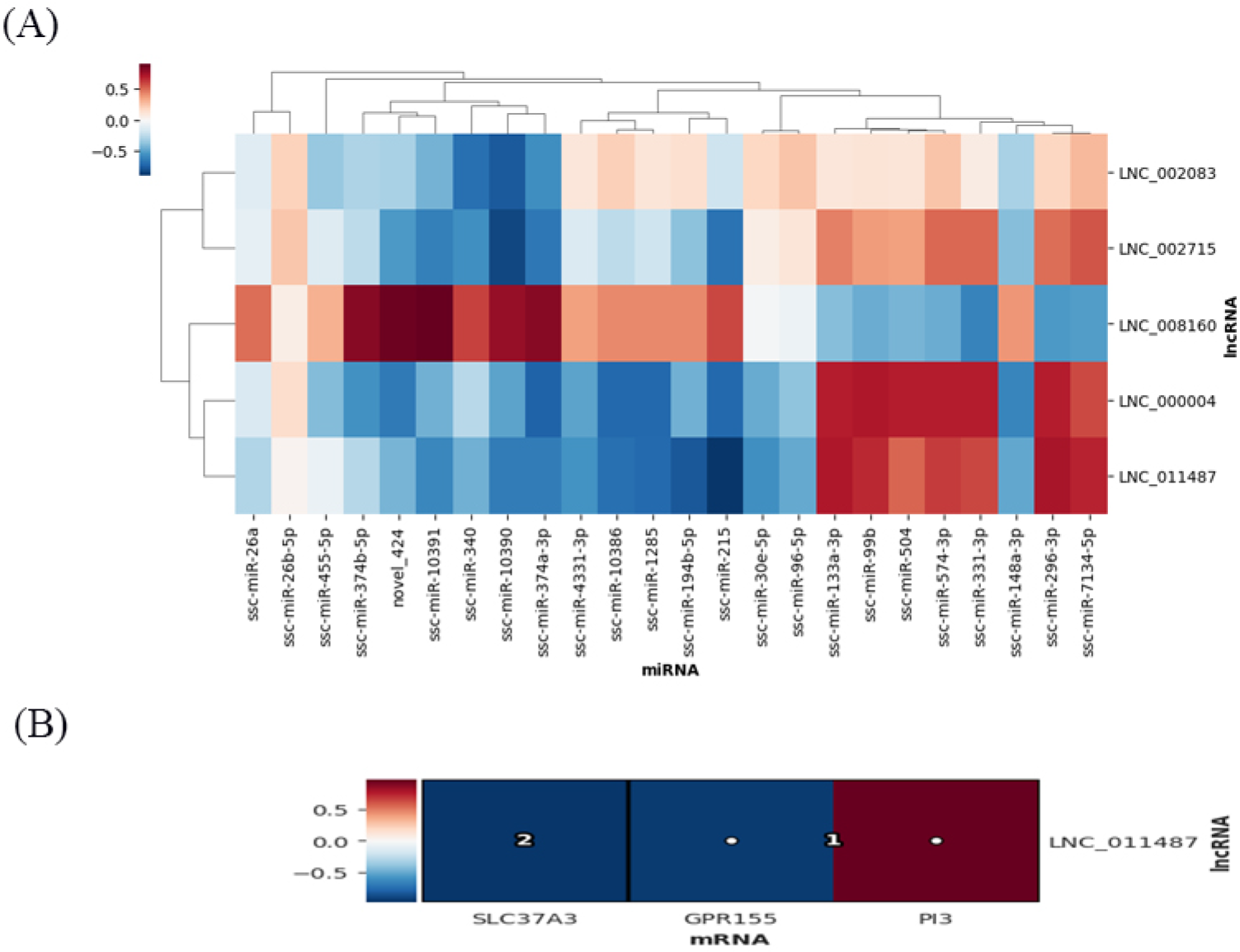
2.6. Multi−Omics Association Analysis
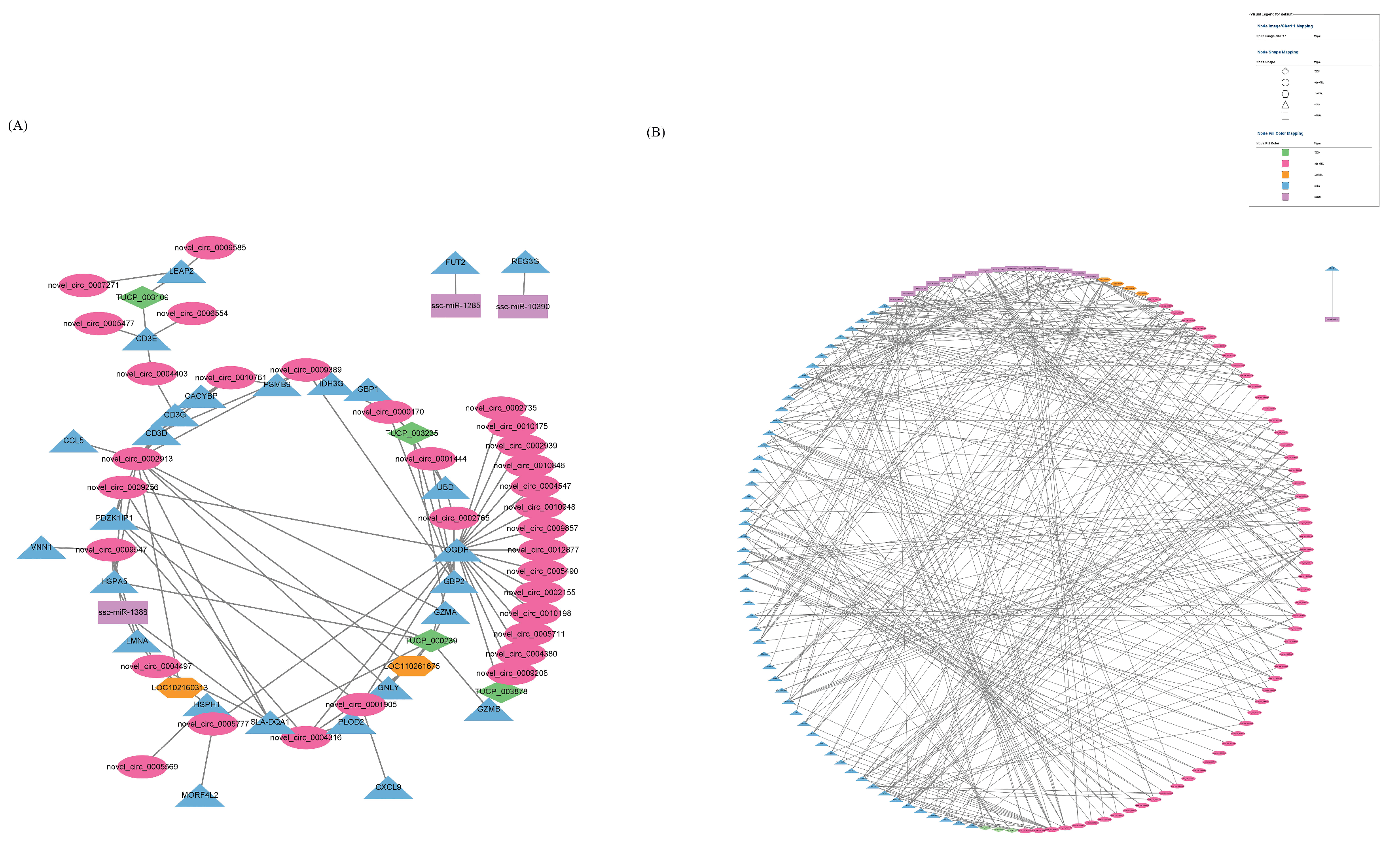
2.7. Construction of Differentially Expressed RNA Association Network
3. Results
3.1. Expression Profiles of Differentially Expressed mRNAs
3.2. Expression Profiles of Differentially Expressed miRNAs
3.3. Expression Profiles of Differentially Expressed cicrRNAs
3.4. Expression Profiles of Differentially Expressed lncRNAs
3.5. Expression Profiles of Differentially Expressed TUCP
3.6. Combined Analysis of mRNAs, miRNAs, lncRNAs, TUCP, cicrRNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.-L.; Shi, W.-F.; Zhang, W.; Zhu, Y.; Zhang, Y.-W.; Xie, Q.-M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Chattha, K.S.; Roth, J.A.; Saif, L.J. Strategies for Design and Application of Enteric Viral Vaccines. Annu. Rev. Anim. Biosci. 2015, 3, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, H.; Bi, Z.; Gu, J.; Gong, W.; Luo, S.; Zhang, F.; Song, D.; Ye, Y.; Tang, Y. Complete Genome Sequence of a Novel Swine Acute Diarrhea Syndrome Coronavirus, CH/FJWT/2018, Isolated in Fujian, China, in 2018. Microbiol. Resour. Announc. 2018, 7, e01259-18. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, Q.-Z.; Lu, W.; Yang, Y.-L.; Chen, R.; Huang, Y.-W.; Wang, B. A Comparative Analysis of Coronavirus Nucleocapsid (N) Proteins Reveals the SADS-CoV N Protein Antagonizes IFN-β Production by Inducing Ubiquitination of RIG-I. Front. Immunol. 2021, 12, 688758. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, Y.; Yan, X.; Tang, X.; Li, Q.; Tan, Y.; Lan, T.; Ma, J. Swine acute diarrhea syndrome coronavirus (SADS-CoV) antagonizes interferon-β production via blocking IPS-1 and RIG-I. Virus Res. 2019, 278, 197843. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, Y.; Yan, X.; Tang, X.; Zhou, L.; Li, Q.; Lan, T.; Ma, J. Swine Acute Diarrhea Syndrome Coronavirus Nucleocapsid Protein Antagonizes Interferon-βProduction via Blocking the Interaction Between TRAF3 and TBK1. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, L.; Shi, H.; Feng, S.; Feng, T.; Chen, J.; Zhang, X.; Han, Y.; Liu, J.; Wang, Y.; et al. Swine acute diarrhea syndrome coronavirus replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. Virology 2021, 565, 96–105. [Google Scholar] [CrossRef]
- Edwards, C.E.; Yount, B.L.; Graham, R.L.; Leist, S.R.; Hou, Y.; Dinnon III, K.H.; Sims, A.C.; Swanstrom, J.; Gully, K.; Scobey, T.D.; et al. Swine acute diarrhea syndrome coronavirus replication in primary human cells reveals potential susceptibility to infection. Proc. Natl. Acad. Sci. USA 2020, 117, 26915–26925. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Tan, C.W.; Xie, S.; Chen, Y.; Yao, Y.; Zhao, K.; Zhu, Y.; Wang, Q.; Liu, M.; Yang, X.; et al. Identification of ZDHHC17 as a Potential Drug Target for Swine Acute Diarrhea Syndrome Coronavirus Infection. mBio 2022, 12, e02342-21. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, J.; Luo, Y.; Yan, X.L.; Wu, Z.X.; He, L.L.; Tan, Y.R.; Zhou, Z.H.; Li, Q.N.; Zhou, L.; et al. Attenuation of a virulent swine acute diarrhea syndrome coronavirus strain via cell culture passage. Virology 2019, 538, 61–70. [Google Scholar] [CrossRef]
- Wei, H.Y.; Huang, S.; Wang, J.; Gao, F.; Jiang, J. Comparison of methods for library construction and short read annotation of shellfish viral metagenomes. Genes Genom. 2018, 40, 281–288. [Google Scholar] [CrossRef]
- Linsen, S.E.; de Wit, E.; Janssens, G.; Heater, S.; Chapman, L.; Parkin, R.K.; Fritz, B.; Wyman, S.K.; de Bruijin, E.; Voest, E.E.; et al. Limitations and possibilities of small RNA digital gene expression profiling. Nat. Methods 2009, 6, 474–476. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Huang, D.; Meng, J.; Wei, Z. Detecting RNA modification using direct RNA sequencing: A systematic review. Comput. Struct. Biotechnol. J. 2022, 20, 5740–5749. [Google Scholar] [CrossRef]
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Young, M.D.; Davidson, N.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Goseq: Gene Ontology testing for RNA-seq datasets. R Bioconductor 2012, 8, 1–25. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, A.R.; Sucipto, K.; Rahnavard, A.; Franzosa, E.A.; Mclver, L.J.; Lloyd-Price, J.; Schwager, E.; Weingart, G.; Moon, Y.S.; Morgan, X.C.; et al. High-sensitivity pattern discovery in large, paired multiomic datasets. Bioinformatics 2022, 38 (Suppl. S1), i378–i385. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Y.; Ding, X.; Li, W. Identification of lncRNA/circRNA-miRNA-mRNA ceRNA Network as Biomarkers for Hepatocellular Carcinoma. Front. Genet. 2022, 13, 838869. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.; Grinstein, S. Phagocytosis and innate immunity. Curr. Opin. Immunol. 2002, 14, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hu, Q.; Zang, X.; Zhou, C.; Liu, D.; Liu, G.; Hong, L. Analysis of Transcripts of Uncertain Coding Potential Using RNA Sequencing During the Preattachment Phase in Goat Endometrium. DNA Cell Biol. 2021, 40, 998–1008. [Google Scholar] [CrossRef]
- Zhu, X.H.; Han, L.; Zhang, R.; Zhang, P.; Chen, F.; Yu, J.; Luo, H.; Han, X. The functional activity of donor kidneys is negatively regulated by microribonucleic acid-451 in different perfusion methods to inhibit adenosine triphosphate metabolism and the proliferation of HK2 cells. Bioengineered 2022, 13, 12706–12717. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Watanabe, O.; Nakamura, M.; Yamamura, T.; Ando, T.; Goto, H.; Hirooka, Y. S100G expression and function in fibroblasts on colitis induction. Int. Immunopharmacol. 2016, 39, 92–96. [Google Scholar] [CrossRef] [PubMed]
- ROCK2 agent stops GVHD. Nat. Biotechnol. 2021, 39, 1323. [CrossRef]
- Zhou, C.; Liang, Y.; Zhou, L.; Yan, Y.; Liu, N.; Zhang, R.; Huang, Y.; Wang, M.; Yang, Y.; Ali, D.W.; et al. TSPAN1 promotes autophagy flux and mediates cooperation between WNT-CTNNB1 signaling and autophagy via the MIR454-FAM83A-TSPAN1 axis in pancreatic cancer. Autophagy 2021, 17, 3175–3195. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Latorre, J.; Lluch, A.; Ortega, F.J.; Comas, F.; Arnoriaga-Rodriguez, M.; Ricart, W.; Fernadez-Real, J.M. Lysozyme is a component of the innate immune system linked to obesity associated-chronic low-grade inflammation and altered glucose tolerance. Clin. Nutr. 2021, 40, 1420–1429. [Google Scholar] [CrossRef]
- Ferrer-Admetlla, A.; Sikora, M.; Laayouri, H.; Esteve, A.; Roubinet, F.; Blancher, A.; Calafell, F.; Bertranpetit, J.; Casals, F. A Natural History of FUT2 Polymorphism in Humans. Mol. Biol. Evol. 2009, 26, 1993–2003. [Google Scholar] [CrossRef]
- Tong, M.; McHardy, I.; Ruegger, P.; Goudarzi, M.; Kashyap, P.C.; Haritunians, T.; Li, X.; Graeber, T.G.; Schwager, E.; Huttenhower, C.; et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 2014, 8, 2193–2206. [Google Scholar] [CrossRef]
- Maroni, L.; Hohenester, S.D.; van de Graaf, S.F.J.; Tolenaars, D.; van Lienden, K.; Verheij, J.; Marzioni, M.; Karisen, T.H.; Elferink, R.P.J.O.; Beuers, U. Knockout of the primary sclerosing cholangitis-risk gene Fut2 causes liver disease in mice. Hepatology 2017, 66, 542–554. [Google Scholar] [CrossRef]
- Choi, J.-H.; Jo, H.S.; Lim, S.; Kim, H.-T.; Lee, K.W.; Moon, K.H.; Ha, T.; Kwak, S.S.; Kim, Y.; Lee, E.J.; et al. mTORC1 accelerates retinal development via the immunoproteasome. Nat. Commun. 2018, 9, 2502. [Google Scholar] [CrossRef] [Green Version]
- Schall, T.J.; Jongstra, J.; Dryer, B.J.; Jorgensen, J.; Clayberger, C.; Davis, M.M.; Krensky, A.M. A human T cell-specific molecule is a member of a new gene family. J. Immunol. 1988, 141, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Gensollen, T.; Bourges, C.; Rihet, P.; Rostan, A.; Millet, V.; Noguchi, T.; Bourdon, V.; Sobol, H.; Dubuquoy, L.; Bertin, B.; et al. Functional Polymorphisms in the Regulatory Regions of the VNN1 Gene Are Associated with Susceptibility to Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2013, 19, 2315–2325. [Google Scholar] [CrossRef]
- De Oliveira, A.P.; Lopes, A.L.F.; Pacheco, G.; de Sá Guimarães Nolêto, I.R.; Nicolau, L.A.D.; Medeiros, J.V.R. Premises among SARS-CoV-2, dysbiosis and diarrhea: Walking through the ACE2/mTOR/autophagy route. Med. Hypotheses 2020, 144, 110243. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Patel, K.K.; Komatsu, M.; Virgin, H.W.; Stappenbeck, T.S. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009, 5, 250–252. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Wumesh, K.C.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2009, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Chisari, F.V. Viruses and the autophagy machinery. Cell Cycle 2010, 9, 1295–1307. [Google Scholar] [CrossRef]
- Rivas-Fuentes, S.; García-García, E.; Nieto-Castañeda, G.; Rosales, C. Fcγ receptors exhibit different phagocytosis potential in human neutrophils. Cell. Immunol. 2010, 263, 114–121. [Google Scholar] [CrossRef]
- Kang, J.; Park, K.-H.; Kim, J.-J.; Jo, E.-K.; Han, M.-K.; Kim, U.-H. The Role of CD38 in Fcγ Receptor (FcγR)-mediated Phagocytosis in Murine Macrophages. J. Biol. Chem. 2012, 287, 14502–14514. [Google Scholar] [CrossRef]
- Zhang, F.; Yuan, W.; Li, Z.; Zhang, Y.; Ye, Y.; Li, K.; Ding, Z.; Chen, Y.; Cheng, T.; Wu, Q.; et al. RNA-Seq-Based Whole Transcriptome Analysis of IPEC-J2 Cells During Swine Acute Diarrhea Syndrome Coronavirus Infection. Front. Vet. Sci. 2020, 7, 492. [Google Scholar] [CrossRef]
- Tezuka, H.; Ohteki, T. Regulation of IgA Production by Intestinal Dendritic Cells and Related Cells. Front. Immunol. 2019, 10, 1891. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Wang, Z.; Wang, X.; Zhan, W.; Wu, D. OGDH is involved in sepsis induced acute lung injury through the MAPK pathway. J. Thorac. Dis. 2021, 13, 5042–5054. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Tang, X.; Zhou, L.; Lv, X.; Gao, L.; Lan, T.; Sun, Y.; Ma, J. Combined Analysis of the Whole Transcriptome of Piglets Infected with SADS−CoV Virulent and Avirulent Strains. Microorganisms 2023, 11, 409. https://doi.org/10.3390/microorganisms11020409
Li Q, Tang X, Zhou L, Lv X, Gao L, Lan T, Sun Y, Ma J. Combined Analysis of the Whole Transcriptome of Piglets Infected with SADS−CoV Virulent and Avirulent Strains. Microorganisms. 2023; 11(2):409. https://doi.org/10.3390/microorganisms11020409
Chicago/Turabian StyleLi, Qianniu, Xiaoyu Tang, Ling Zhou, Xiaocheng Lv, Long Gao, Tian Lan, Yuan Sun, and Jingyun Ma. 2023. "Combined Analysis of the Whole Transcriptome of Piglets Infected with SADS−CoV Virulent and Avirulent Strains" Microorganisms 11, no. 2: 409. https://doi.org/10.3390/microorganisms11020409