
Cloning and Molecular Characterization of the Recombinant CVB4E2 Immunogenic Viral Protein (rVP1), as a Potential Subunit Protein for Vaccine and Immunodiagnostic Reagent Candidate

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral Strain, Bacterial Strain, Plasmid and Growth Medium
2.2. PCR Amplification, Cloning and Sequencing of the CVB4 VP1 Gene
2.3. Preparation of Cell Lysates for SDS-PAGE
2.4. SDS-PAGE and Western Blot Analysis of rVP1 Subunit Protein
2.5. Purification of rVP1 Subunit Protein
2.6. Bioinformatic Analysis
3. Results
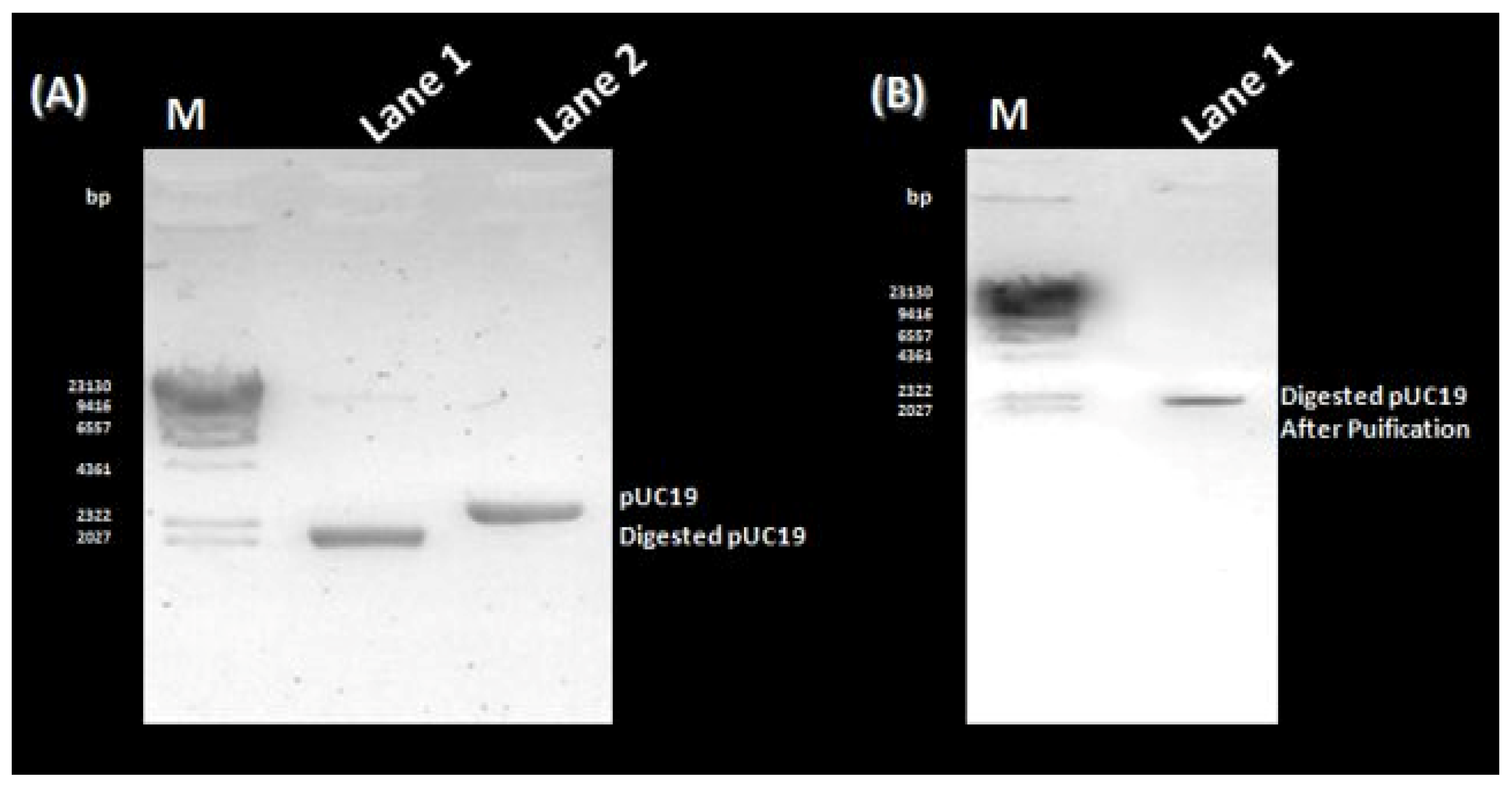
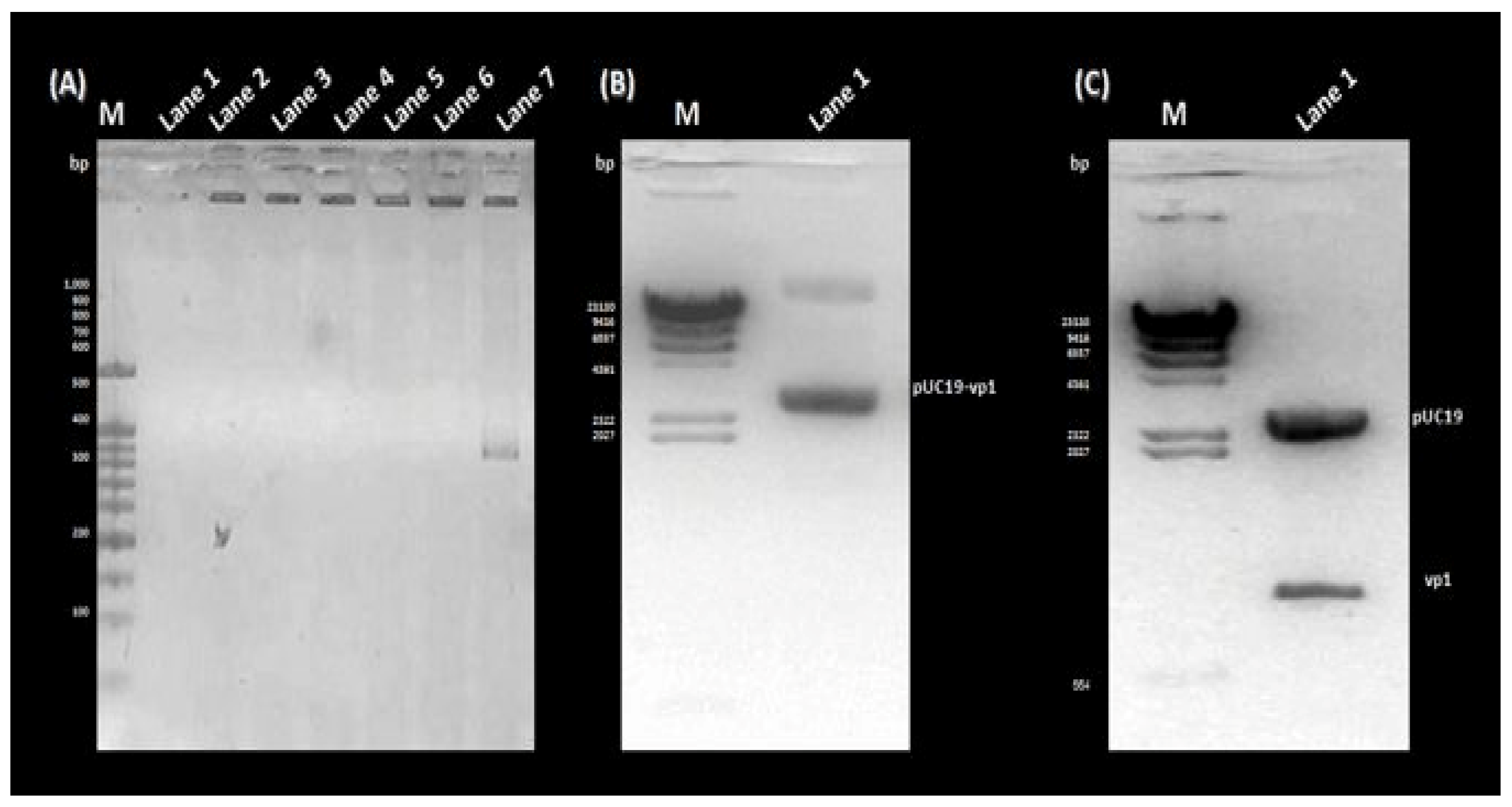
3.1. PCR Amplification, Cloning, and Sequencing of the rVP1 Gene
3.2. SDS-PAGE Analysis, Western Blotting and Purification of the rVP1 Subunit Protein
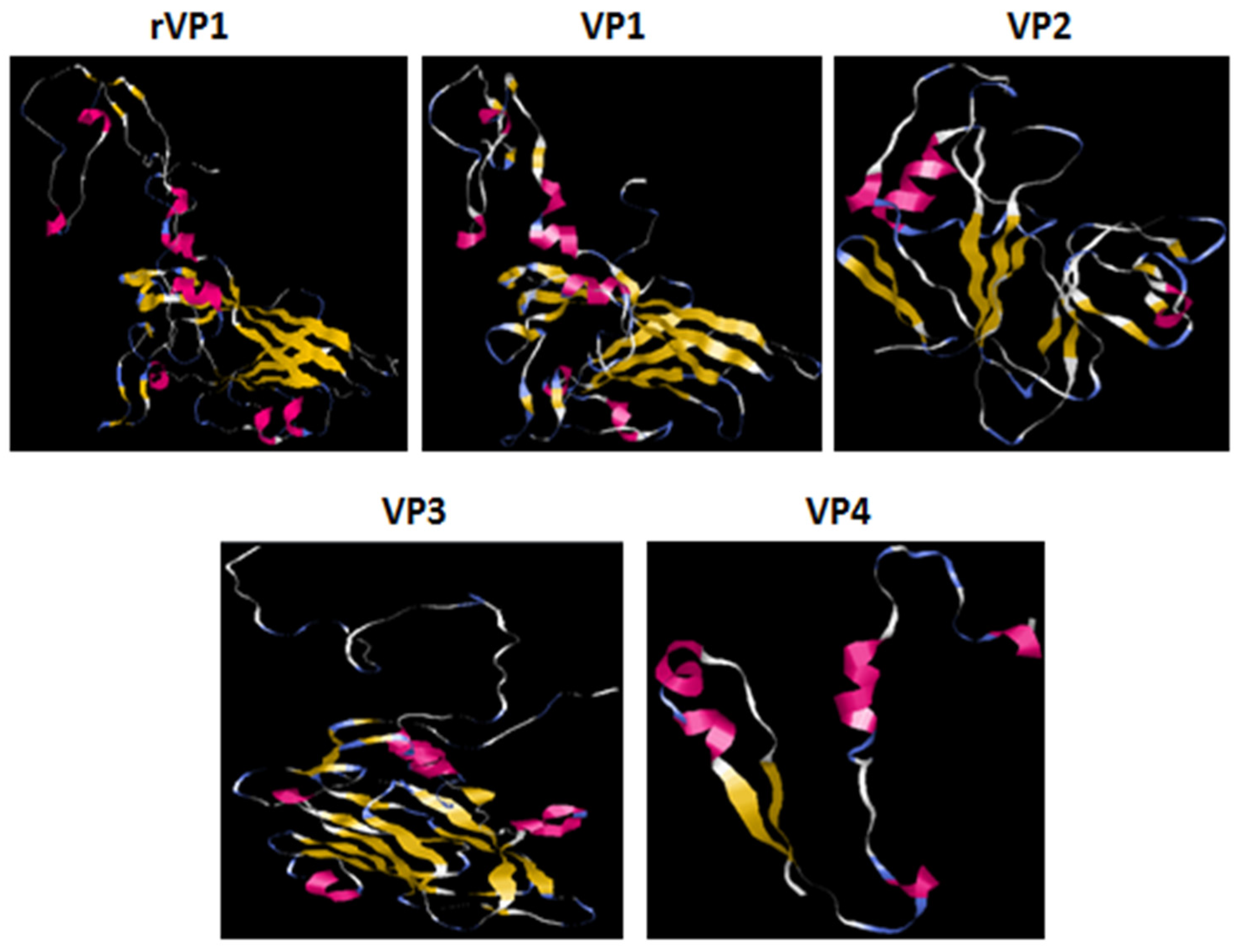
3.3. Bioinformatic Analysis of the Sequence and Structure of the rVP1 Subunit Protein
3.3.1. Bioinformatic Analysis of the Nucleotide Sequence of rVP1
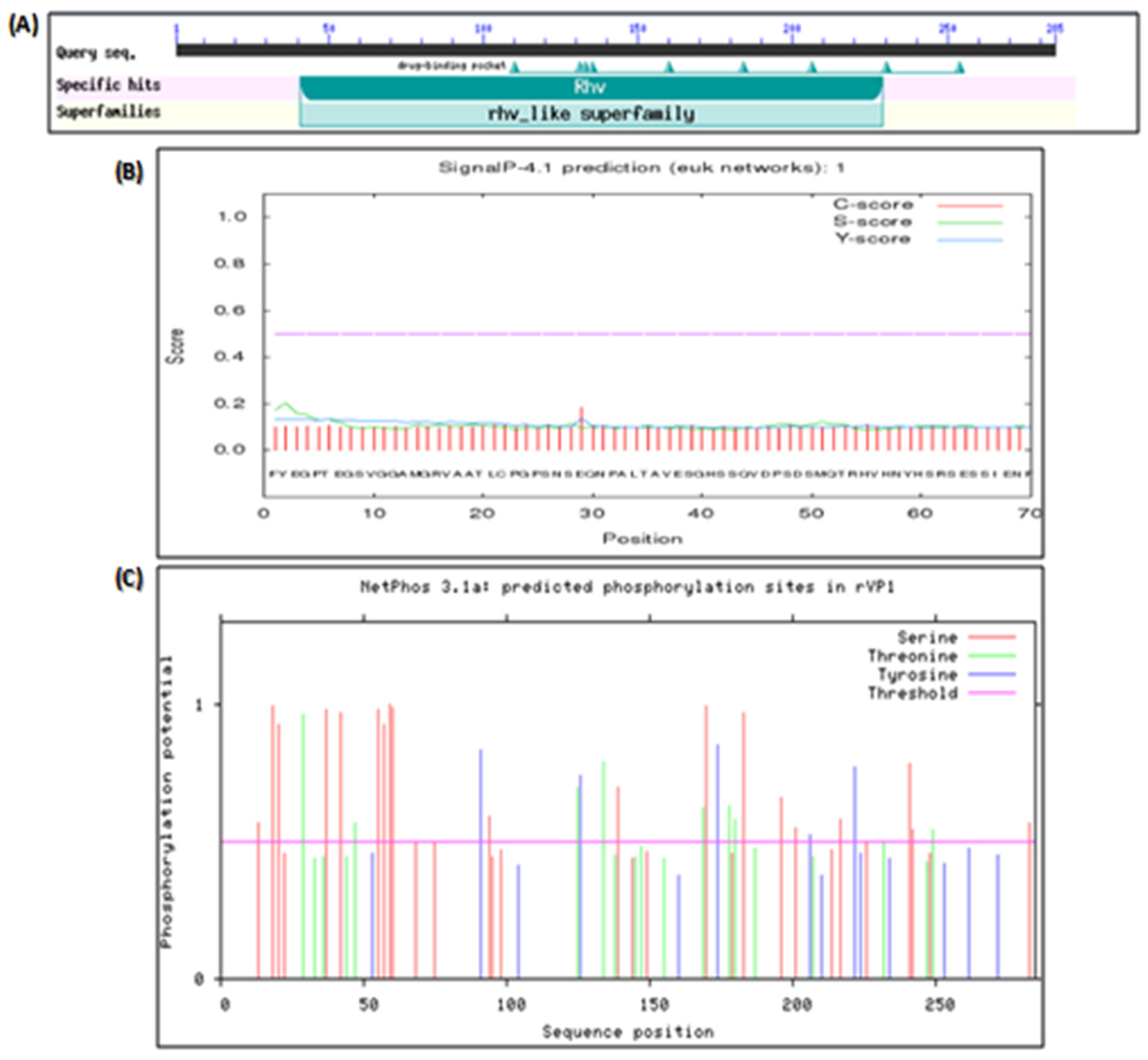
3.3.2. Bioinformatic Analysis of the Polypeptide Sequence of rVP1
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV virus taxonomy profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Solomon, T.; Lewthwaite, P.; DaCardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Hassine, I.H.; Gharbi, J.; Hamrita, B.; Almalki, M.A.; Rodríguez, J.F.; Ben M’hadheb, M. Characterization of Coxsackievirus B4 virus-like particles VLP produced by the recombinant baculovirus-insect cell system expressing the major capsid protein. Mol. Biol. Rep. 2020, 47, 2835–2843. [Google Scholar] [CrossRef]
- Ben M’hadheb, M.; Souii, A.; Harrabi, M.; Jrad-Battikh, N.; Gharbi, J. In Vitro-reduced translation efficiency of coxsackievirus B3 Sabin3-like strain is correlated to impaired binding of cellular initiation factors to viral IRES RNA. Curr. Microbiol. 2015, 70, 756–761. [Google Scholar] [CrossRef]
- Gharbi, J.; Hiar, R.; Ben M’hadheb, M.; Jaïdane, H.; Bouslama, L.; N’saïbia, S. Aouni, M. Nucleotide sequences of IRES domains IV and V of natural ECHO virus type 11 isolates with different replicative capacity phenotypes. Virus Genes 2006, 32, 269–276. [Google Scholar] [CrossRef]
- Acharya, R.; Fry, E.; Stuart, D.; Fox, G.; Rowlands, D.; Brown, F. The three-dimensional structure of foot-and-mouth disease virus at 2.9 Å resolution. Nature 1989, 337, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Flemister, M.R.; Brown, B.A.; Pallansch, M.A. Typing of human enteroviruses by partial sequencing of VP1. J. Clin. Microbiol. 1999, 37, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Austin, M.; Onodera, T.; Notkins, A.L. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 1979, 300, 1173–1179. [Google Scholar] [CrossRef]
- Hoover, D.M.; Lubkowski, J. DNAWorks: An automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic. Acids. Res. 2002, 30, 1–7. [Google Scholar] [CrossRef]
- Niu, X.N.; Wei, Z.Q.; Zou, H.F.; Xie, G.G.; Wu, F.; Li, K.J.; Jiang, W.; Tang, J.L.; He, Y.Q. Complete sequence and detailed analysis of the first indigenous plasmid from Xanthomonas oryzae pv. Oryzicola. BMC Microbiol. 2015, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Messing, J.; Vieira, J. A new pair of M13 vectors for selecting either DNA strand of double-digest restriction fragments. Gene 1982, 19, 269–276. [Google Scholar] [CrossRef]
- Messing, J. New M13 vectors for cloning. Methodes Enzymol. 1983, 101, 20–78. [Google Scholar]
- Yanisch-perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 1983, 27, 493–497. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.T.; Niemela, S.L.; Miller, R.H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. USA 1989, 86, 2172–2175. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy server. In The proteomics Protocols Handbook; John, M.W., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. Signal P 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Blom, N.S.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef]
- Selbig, J.; Mevissen, T.; Lengauer, T. Decision tree-based formation of consensus protein secondary structure prediction. Bioinformatics 1999, 15, 1039–1046. [Google Scholar] [CrossRef]
- Idicula-thomas, S.; Balaji, P.V. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on overexpression in Escherichia coli. Protein Sci. 2005, 14, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Whitney, K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid. Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rVP1 | VP1 | VP2 | VP3 | VP4 | |
|---|---|---|---|---|---|
| SOPMA | |||||
| α-helix | 23.47% | 20.07% | 17.34% | 17.15% | 17.56% |
| β-folding | 25.63% | 23.24% | 26.61% | 29.29% | 22.06% |
| β-turns | 3.25% | 2.11% | 7.66% | 5.44% | 4.41% |
| Random coils | 47.65% | 54.58% | 48.39% | 48.12% | 55.88% |
| Predictprotein | |||||
| α-helix | 4.69% | 8.45% | 2.82% | 8.37% | 13.24% |
| β-folding | 23.47% | 26.41% | 24.60% | 23.43% | 16.18% |
| Random coils | 71.84% | 65.14% | 72.58% | 68.20% | 70.59% |
| Exposed AA | 47.65% | 45.42% | 43.95% | 42.68% | 16.18% |
| Buried AA | 7.22% | 7.04% | 6.05% | 11.30% | - |
| Intermediate AA | 45.13% | 47.54% | 50% | 46.03% | 83.82% |
| Template | GMQE | QMEAN | |
|---|---|---|---|
| rVP1 | 1h8t.1A | 0.85 | −3.29 |
| VP1 | 4GB3 | 0.78 | −3.06 |
| VP2 | 1mqt.1.B | 0.57 | −6.83 |
| VP3 | 1oop.1.C | 0.93 | −1.90 |
| VP4 | 1pov.1.A | 0.57 | −5.03 |
| Capsid | N° | Start | End | Peptide | Length |
|---|---|---|---|---|---|
| rVP1 | 1 | 7 | 7 | E | 1 |
| 2 | 18 | 63 | AATLCPGPSNSEQNPALTAVESGHSSQVDPSDSMQTRHVHNYHSRS | 46 | |
| 3 | 83 | 93 | SSAESNNLKRY | 11 | |
| 4 | 30 | 40 | QEMSSASNSDV | 11 | |
| 5 | 155 | 168 | PVPTSVNDYVWQTS | 13 | |
| 6 | 202 | 213 | WSNFSRDGIYGY | 12 | |
| 7 | 230 | 234 | SSPGG | 5 | |
| 8 | 258 | 273 | LCQYKKAKNGNFDVEA | 16 | |
| VP1 | 1 | 5 | 31 | ESVERAMGRVADTIARGPSNSEQIPAL | 27 |
| 2 | 33 | 50 | AVETGHTSQVDPSDTMQT | 18 | |
| 3 | 53 | 53 | V | 1 | |
| 4 | 55 | 61 | NYHSRSE | 7 | |
| 5 | 82 | 96 | AESNNLKRYAEWVIN | 15 | |
| 6 | 127 | 138 | QEMSTATNSVVP | 12 | |
| 7 | 152 | 165 | PVPTSVNDYVWQTS | 14 | |
| 8 | 176 | 176 | N | 1 | |
| 9 | 199 | 209 | WSNFSRDGIYG | 11 | |
| 10 | 227 | 231 | SSPGGL | 6 | |
| 11 | 251 | 280 | PPRRLCQYKKAKNVNFDVEAVTTERASLVTT | 31 | |
| VP2 | 1 | 5 | 27 | EECGYSDRVRSITLGNSTITTQE | 13 |
| 2 | 38 | 60 | WPDYLSDEEATAEDQPTQPDVAT | 23 | |
| 3 | 66 | 92 | LKSVKWRCSQRGGGGSSQMRCQRWVSL | 27 | |
| 4 | 130 | 165 | WGALTRKMRPHMVIYAEGKQQNNLNKMQSQVRLLCK | 36 | |
| 5 | 213 | 215 | SHY | 3 | |
| 6 | 223 | 244 | RERPPTFPSLQLLQALNTMACD | 22 | |
| VP3 | 1 | 7 | 39 | TPGSTQFLTSDDFQSPSAMPQFDVNPEMNIPGR | 33 |
| 2 | 55 | 67 | INNLQANLKTMEA | 13 | |
| 3 | 72 | 80 | VRSTDEMGQ | 9 | |
| 4 | 91 | 94 | SSVL | 4 | |
| 5 | 139 | 150 | GAGAPDSRKNAM | 12 | |
| 6 | 202 | 206 | PAEAQ | 5 | |
| 7 | 228 | 236 | DTQFIKQDT | 9 | |
| VP4 | 1 | 5 | 19 | STQKTGAHETSLSAT | 15 |
| 2 | 29 | 54 | INYYKDAASNSANRQDFTQDPSKFTE | 26 | |
| 3 | 61 | 62 | IK | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadj Hassine, I.; Gharbi, J.; Amara, I.; Alyami, A.; Subei, R.; Almalki, M.; Hober, D.; M’hadheb, M.B. Cloning and Molecular Characterization of the Recombinant CVB4E2 Immunogenic Viral Protein (rVP1), as a Potential Subunit Protein for Vaccine and Immunodiagnostic Reagent Candidate. Microorganisms 2023, 11, 1192. https://doi.org/10.3390/microorganisms11051192
Hadj Hassine I, Gharbi J, Amara I, Alyami A, Subei R, Almalki M, Hober D, M’hadheb MB. Cloning and Molecular Characterization of the Recombinant CVB4E2 Immunogenic Viral Protein (rVP1), as a Potential Subunit Protein for Vaccine and Immunodiagnostic Reagent Candidate. Microorganisms. 2023; 11(5):1192. https://doi.org/10.3390/microorganisms11051192
Chicago/Turabian StyleHadj Hassine, Ikbel, Jawhar Gharbi, Imene Amara, Ameera Alyami, Reem Subei, Mohammed Almalki, Didier Hober, and Manel Ben M’hadheb. 2023. "Cloning and Molecular Characterization of the Recombinant CVB4E2 Immunogenic Viral Protein (rVP1), as a Potential Subunit Protein for Vaccine and Immunodiagnostic Reagent Candidate" Microorganisms 11, no. 5: 1192. https://doi.org/10.3390/microorganisms11051192