
RNase Y Autoregulates Its Synthesis in Bacillus subtilis

1
Expression Génétique Microbienne, Institut de Biologie Physico-Chimique, CNRS, Université Paris Cité, 75005 Paris, France
2
Inovarion, 75005 Paris, France
*
Author to whom correspondence should be addressed.
Microorganisms 2023, 11(6), 1374; https://doi.org/10.3390/microorganisms11061374
Submission received: 7 April 2023
/
Revised: 18 May 2023
/
Accepted: 22 May 2023
/
Published: 24 May 2023
(This article belongs to the Special Issue 10th Anniversary of Microorganisms: Past, Present and Future)
Abstract
:The instability of messenger RNA is crucial to the control of gene expression. In Bacillus subtilis, RNase Y is the major decay-initiating endoribonuclease. Here, we show how this key enzyme regulates its own synthesis by modulating the longevity of its mRNA. Autoregulation is achieved through cleavages in two regions of the rny (RNase Y) transcript: (i) within the first ~100 nucleotides of the open reading frame, immediately inactivating the mRNA for further rounds of translation; (ii) cleavages in the rny 5′ UTR, primarily within the 5′-terminal 50 nucleotides, creating entry sites for the 5′ exonuclease J1 whose progression is blocked around position −15 of the rny mRNA, potentially by initiating ribosomes. This links the functional inactivation of the transcript by RNase J1 to translation efficiency, depending on the ribosome occupancy at the translation initiation site. By these mechanisms, RNase Y can initiate degradation of its own mRNA when the enzyme is not occupied with degradation of other RNAs and thus prevent its overexpression beyond the needs of RNA metabolism.
1. Introduction
Control of gene expression requires the instability of messenger RNA. In bacteria, mRNA half-lives vary from seconds to over an hour. The decay of mRNA generally follows an “all-or-none” pattern, implying that if control is to be efficient, it must occur at the rate-limiting initial step of the degradation process. Studies in Escherichia coli and Bacillus subtilis, two evolutionary very distant organisms, have provided essential knowledge on the initiation of mRNA decay by endonucleolytic cleavage that can be summarized by “different enzymes—similar strategies”. The crucial role of the endoribonucleases RNase E (E. coli) and RNase Y (B. subtilis) in producing short-lived decay intermediates is now clearly recognized [1]. In B. subtilis, RNase Y affects global mRNA stability by cleaving its substrates with an RNase E-like single strand-specific cleavage specificity [2]. The intracellular levels of a majority of transcripts are affected in RNase Y-depleted strains [3,4,5]. An alternative degradation pathway has been described for some mRNAs where the decay of the primary transcript is triggered by the RNA pyrophosphohydrolase RppH which generates a monophosphorylated intermediate that becomes vulnerable to 5′-exonucleolytic digestion by RNase J1 [6]. RNase J, comprising RNase J1 and J2 subunits, is a multifunction enzyme with both endonuclease and 5′ exonuclease activity; the latter activity being essentially a property of the J1 subunit that requires a monophosphorylated 5′-end (or 5′-OH) [7,8,9,10]. RNase J1 thus functions primarily in the 5′ exonucleolytic degradation of RNA decay intermediates generated by endonucleolytic cleavage or 5′-end conversion. Interestingly, a monophosphorylated RNA 5′ end also potentiates the endonucleolytic activity of RNase Y in vitro [2], but it is unknown whether this plays a significant role in the RNase Y-dependent degradation of RNAs in vivo.
The sequence specificity of RNase Y in B. subtilis and other Gram-positive organisms is rather loose with a preference for UA-rich single-stranded sequences often flanked by secondary structures [2,11,12,13,14]. The same description also holds for RNase E cleavage sites with a few differences in the proposed sequence context [15,16,17,18,19,20]. E. coli RNase E can effectively replace RNase Y in B. subtilis, enabling a surprising reversal of transcript profiles not only of individual genes but also on a genome-wide scale [21].
Other important features shared between RNase Y and RNase E include a pseudo-compartmentalization; both enzymes are tethered to the inner cell membrane [22,23], in the case of RNase Y, via a single membrane-traversing N-terminal helix [24], (RNase Y is called YmdA in this reference). This is coherent with the predominant distribution of translating ribosomes around the cell periphery [25,26]. RNase Y diffuses rapidly across the membrane in the form of dynamic short-lived foci that become more abundant and increase in size following transcription arrest [27]. A degradosome complex centered on RNase Y has been proposed [28,29] but, unlike the RNase E-based degradosome in E. coli [30,31], cannot be isolated in the absence of crosslinking agents. Whether RNase Y can interact with other ribonucleases in any meaningful way remains an open question [32,33,34]. However, three small proteins, YlbF, YmcA, and YaaT forming the so-called Y-complex [35,36] were shown to alter RNase Y activity in vivo [37] by affecting the abundance of certain riboswitches and favoring the efficient maturation of operon mRNAs [13]. These effects of the Y-complex might be achieved by shifting the assembly status of RNase Y towards fewer and smaller membrane tethered complexes [21].
As the key ribonuclease initiating mRNA decay in B. subtilis, there is a need to keep the activity of RNase Y in a tightly controlled steady supply. Here, we show that RNase Y can cleave its own mRNA at several positions, within the 5′UTR and the open reading frame. This homeostatic mechanism allows tight regulation of its synthesis by modulating the decay rate of the rny (RNase Y) mRNA in response to changes in cellular RNase Y activity.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
The B. subtilis strains used in this work are derivatives of strain SSB1002, a wild-type laboratory stock strain derived from strain 168. E. coli strains JM109 [38] and XL1-Blue (Stratagene) were used for plasmid constructions and site-directed mutagenesis experiments. E. coli recA+ strain JM101 [39] was used for plasmid purification before transformation into B. subtilis cells. B. subtilis and E. coli strains were grown at 37 °C in LB medium with aeration to mid-log phase (OD600~0.5–0.6). Strains carrying the replicative control plasmid pWH353m (pC) or its rny-expressing pHMD22 (pY) or pHMD33 (pY*) derivative plasmids were grown without or with inducer (0.2 μg/mL of tetracycline). Antibiotics were added, at the following concentrations: ampicillin (100 µg/mL), kanamycin (5 µg/mL), chloramphenicol (5 µg/mL), neomycin (7 µg/mL), and spectinomycin (100 μg/mL) for B. subtilis, and ampicillin (200 μg/mL) and kanamycin (100 μg/mL) for E. coli.
Translational rny-lacZ fusions (see plasmids) are based on plasmid pHM14 [40] and were integrated in single copy at the amyE locus of the B. subtilis SSB1002 strain (see Table 1). Insertions were confirmed by PCR and sequencing. For the long rny-lacZ fusion and its mutated variants, the products of the ligation reaction were amplified with primers HP2248 and HP2249 and used to transform SSB1002 directly. Primers used in this study are listed in Table 2.
A rny deletion (∆rny::spc) strain was constructed by transforming B. subtilis wild-type and derivative strains with a DNA cassette composed of the spectinomycin resistance gene (spc) flanked by ~1 kb sequences corresponding to the up- and downstream regions of the rny ORF on the chromosome. The final fragment was obtained by overlapping PCR of three individual fragments. The 5′ and 3′ flanking regions were amplified using primer pairs HP2078-HP2080 and HP2104Rev-HP2083, respectively, the spc cassette was amplified from plasmid pDG1727 [41] with primers HP2079-HP2104. A fragment composed of the spc cassette and the 3′ region of rny was amplified with primers HP2079-HP2083 using the individual overlapping fragments. Finally, the full-length DNA cassette was amplified with oligonucleotides HP2078-HP2083 and two corresponding fragments from the first and second step as the DNA template.
The rnjA deletion strain SSB1086 (∆rnjA::tet) was constructed by transforming strain SSB525 with plasmid pHMJ26 which replaces the rnjA ORF with the tetracycline resistance cassette by double cross-over recombination.
2.2. Plasmid Constructs
pHMD17. Plasmid pHMD17 contains the entire rny promoter region with the rny ORF up to codon 5 inserted as a BamHI-EcoRI fragment (primers HP1714-HP1715) in-frame into the lacZ fusion vector pHM14 [40].
pHMD22. Plasmid pHMD22 contains the rny gene including the Shine–Dalgarno sequence as a 1.6 kb PCR fragment (primers HP1832-HP1833) inserted into the inducible expression vector pWH353m between sites SphI and SalI.
pHMD28. Plasmid pHMD28 contains the entire rny promoter region with the rny ORF up to codon 13 inserted as a BamHI-EcoRI fragment (primers HP2164-HP2165) in-frame into the lacZ fusion vector pHM14 [40].
pHMD29. Plasmid pHMD29 contains the entire rny promoter region with the rny ORF up to codon 31 inserted as a BamHI-EcoRI fragment (primers HP1714-HP2166) in-frame into the lacZ fusion vector pHM14 [40].
pHMD33. Plasmid pHMD33 is similar to pHMD22 but contains a mutated rny gene which codes for an inactive RNase Y (mutations H368A and D369A in HD domain). Two overlapping PCR fragments (primer pairs HP1832-HP2228 and HP2227-HP1833) containing the rny double mutations at the overlap were amplified from chromosomal DNA using KOD DNA polymerase (Novagen). Both fragments were used as the template for amplification with primers HP1832-HP1833. The resulting fragment was digested with SalI and SphI and ligated into the respective sites of plasmid pWH353.
pHMD37. Plasmid pHMD37 contains the entire rny promoter region with the rny ORF up to codon 210 inserted as a BamHI-EcoRI fragment (primers HP1714-HP1801) in-frame into the lacZ fusion vector pHM14 [40]. The ligation mix was amplified with primers HP2248-HP2249 and the resulting fragment used for transformation.
pHMD38. Plasmid pHMD38 is similar to pHMD37 but lacks a large part of the rny 5′ UTR (deletion between positions −20 and −158). A PCR fragment (primers HP1714-HP2269) was amplified and used as a megaprimer in a second PCR reaction with primer HP1801 using plasmid pHMD37 as the template. The resulting fragment was inserted as a BamHI-EcoRI fragment in-frame into the lacZ fusion vector pHM14 [40].
pHMD39. Plasmid pHMD38 is similar to pHMD17 but lacks a large part of the rny 5′ UTR (deletion between positions −20 and −158). A PCR fragment (primers HP1714-HP2268) was amplified plasmid pHMD17 as the template and inserted as a BamHI-EcoRI fragment in-frame into the lacZ fusion vector pHM14 [40].
pHMJ26. Plasmid pHMJ26 contains 3 fragments in the following order cloned into the vector pJRD184 [42] between the sites EcoRI and XbaI: a 0.7 kb EcoRI-PstI PCR fragment (primers HP899-HP1024) containing sequences upstream of rnjA, a 2.1 kb PstI-XhoI fragment of plasmid pDG1515 [41] containing the tetracycline resistance cassette and a 0.7 kb XhoI-XbaI PCR fragment (primers HP1039-HP1040) containing sequences downstream of rnjA.
2.3. Western Blot
For Western blot analysis, 20 μg of protein extract prepared as described previously [45] was separated by SDS-PAGE (10%). After electrophoretic transfer of the proteins, the nitrocellulose membrane (GE Healthcare, Chicago, IL, USA) was stained with amido black to check for equal transfer across all lanes. The membrane was blocked for 1 h with 5% skimmed milk in phosphate-buffered saline (PBS)-Tween buffer (100 mM NaH2PO4-Na2HPO4, pH 7.4, 100 mM NaCl, 0.1% Tween) and incubated with a monoclonal RNase Y antibody diluted in PBS-Tween for at least 4 h. Signals were detected by ECL Chemiluminescence (BIO-RAD, ClarityTM Western ECL) associated with a CCD camera (BIO-RAD ChemiDoc XR System+). When necessary, ECL-detected proteins were quantified using the ImageLab software version 6.0.1 (Bio-Rad, Hercules, CA, USA). The uncropped versions of the Western blots are shown in Supplementary Materials.
2.4. Northern Blot
RNA blot analysis was carried out using 5–10 μg of total RNA isolated as described previously [40], separated on a 1% agarose/0.22 M formaldehyde gel, and blotted onto Hybond N+ membranes (GE Healthcare). RNA was crosslinked to the membrane at 120 mJ cm–2 for 1 min. RNA probes were denatured at 80 °C for 3 min, kept on ice for 2 min, and then added to the prehybridized membranes (Amersham Rapid-hyb buffer) for hybridization at 70 °C for 2 h. Membranes were washed at 68 °C with washing solutions I (2 × SSC and 0.1% SDS), II (1 × SSC, 0.1% SDS), and III (0.5 × SSC, 0.1% SDS) for 15 min each. The signals were detected with a GE Healthcare Typhoon FLA 9500 imaging system. As lacZ and 5′ UTR rny probes, T7 RNA polymerase in vitro transcripts were used which were generated from a PCR template obtained with oligonucleotides HP2270-HP2271 and HP2238-HP2239, respectively. Chromosomal DNA of the B. subtilis SSB530 strain was used as a template for PCR. 16S rRNA was used as loading control (hybridization with oligonucleotide HP2113). The uncropped versions of the Northern blots are shown in Supplementary Materials.
2.5. Half-Life Measurements
For half-life measurements, rifampicin (Sigma, St. Louis, MO, USA) was added at 150 µg/mL to cultures at OD600 ~0.5. At 0, 1, 2, 4, 6, and 10 min after addition of rifampicin, 10 mL of cell culture were mixed with 5 mL of frozen buffer (20 mM Tris-HCl pH 7.8, 5 mM MgCl2, 20 mM NaN3), centrifuged, and frozen in dry ice/ethanol. Total RNA from the samples was purified and analyzed by Northern blot as described above.
2.6. Primer Extension Analysis
Primer extension was carried out using 5 μg of total RNA, 5′[32P]-labelled primer HP905 complementary to sequences at the 5′ end of the lacZ ORF and Superscript III reverse transcriptase (Invitrogen, Waltham, MA, USA) following the instructions of the manufacturer. Reactions were carried out at 55 °C for 10 min.
2.7. β-Galactosidase Measurements
For β-galactosidase measurements, cells were grown in the appropriate medium and cell extracts were prepared from 3 to 5 mL of cell culture. Extracts were used for measurements as described elsewhere [46]. All reported values are based on two to four independent measurements. Error bars represent standard deviation (sd).
3. Results
3.1. Rny-lacZ Expression Is Responsive to RNase Y Levels In Vivo
We first assessed the ability of RNase Y to regulate its own expression using two translational lacZ fusions. Both contained the entire 164 nt 5′ UTR of the rny gene identified previously by a transcriptomic analysis [47] and confirmed by primer extension analysis (see below). Transcription is driven by a typical sigma A-type promoter (Figure 1B). The two lacZ constructs differed in the length of the rny ORF fused in-frame to lacZ, i.e., 5 and 210 codons, referred to as short and long fusions, respectively (Figure 1B). The fusions were integrated in single copy at the amyE locus.
Regulated overexpression of RNase Y was achieved by further transforming rny-lacZ-containing strains with the rny expression plasmid pHMD22 that allows transcriptional control of the additional plasmid-borne rny gene by a tetracycline-inducible promoter. Growth in the presence of inducer increased the intracellular RNase Y level up to about 8- to 10-fold (Figure 1C, lower panels, confer lanes 1, 2 and 4, 5). This overexpression of RNase Y only slightly reduced the activity of the short rny-lacZ fusion (~1.3-fold) while the long rny-lacZ fusion was strongly (~6-fold) repressed (Figure 1C, β-gal activities at the top).
To test whether this control requires the catalytic activity of RNase Y we also overproduced, an RNase Y protein where the conserved HD motif in the active site was mutated (H368A/D369A, Figure 1A). We have previously shown that single mutations of the HD motif almost abolish the endonucleolytic activity of RNase Y [2]. In contrast to the wild-type enzyme, overexpressing the inactive form of RNase Y (Figure 1C, lanes 3 and 6) does not decrease the activity of the short nor the long form of the rny-lacZ fusions. On the contrary, expression of the long lacZ fusion was significantly increased under these conditions, suggesting that inactive RNase Y interferes with the activity of the chromosome-encoded wild-type form of the enzyme (Figure 1C, see also Discussion). This increase in β-galactosidase activity is also visible in the Western blot analysis aimed at quantifying RNase Y overproduction (Figure 1C, Western blots at the bottom, cf. lanes 5 and 6). Since the monoclonal antibody is directed against a peptide in the N-proximal 210 aa (but not within the N-terminal 5 aa) of RNase Y (Figure 1A), we could also detect the fusion protein synthesized from the long but not the short form of the rny-lacZ fusion construct (Figure 1C, lower panel, lane 6; see also lower WB panel of Figure 1D).
We next analyzed the effect of the absence of RNase Y on rny-lacZ expression. For this purpose, the entire rny ORF was replaced with a spectinomycin resistance cassette. Expression of the short rny-lacZ fusion remained unchanged in the absence of RNase Y. However, expression of the long fusion was induced 2- to 3-fold when measuring both β-galactosidase activity and the level of fusion protein in the cell by Western blot, respectively (Figure 1D, lanes 9 and 10).
These data indicated that RNase Y might autoregulate its own expression and that control primarily involves rny mRNA sequences between codon 5 and 210.
3.2. RNase Y Affects rny and rny-lacZ Transcript Levels Alike
The data obtained with the short rny-lacZ fusion described above strongly suggested that the lacZ mRNA part of the fusion mRNA does not contribute to autoregulation (see also discussion). Nevertheless, to test how RNase Y affects its own expression, we analyzed the effect of RNase Y on both rny and rny-lacZ mRNA levels. First, we transformed the wild-type strain (SSB1002) with plasmid pHMD22 for inducible RNase Y overexpression (pY) or with the empty vector as control (pC). The rny messenger transcribed from the chromosomal gene was detected with a probe specific for the rny 5′UTR which does not detect rny mRNA transcribed from the plasmid since this gene copy lacked the 5′ UTR. The rny mRNA level transcribed from the chromosome was very sensitive to the intracellular RNase Y concentration (Figure 2A). In the absence of inducer, we observed a two- to three-fold overproduction of RNase Y due to leaky expression. This was already sufficient to strongly reduce chromosomal rny mRNA about four-fold (Figure 2A). In the presence of inducer RNase Y was overproduced ~ten-fold, causing rny mRNA to drop below detectable levels (Figure 2A). In contrast, a similar overproduction of the inactive RNase Y mutant led to a more than two-fold increase in rny transcripts (Figure 2A), mirroring a similar effect on the β-galactosidase expression of the long rny-lacZ fusion under the same conditions (Figure 1C). The transcripts of the short and long rny-lacZ fusions, detected using a lacZ-specific probe, responded in much the same way as the rny mRNA to overproduction of RNase Y (Figure 2B).
While the long fusion mRNA became almost undetectable when overproducing RNase Y, the short fusion mRNA was also reduced two-fold suggesting that RNase Y does not only cleave within the rny ORF but also in the 5′ UTR (Figure 2B). In the absence of RNase Y (Figure 2C), the short rny-lacZ fusion mRNA is barely increased (~1.1-fold) while induction of the long rny-lacZ fusion mRNA is upregulated by a factor of 5-fold compared to a wild-type strain, again suggesting that 5′ proximal sequences in the rny ORF play an important role for controlling the rny transcript levels in the cell. The similar response of rny mRNA and the long rny-lacZ transcript to changes in the cellular level of RNase Y indicated that the regulation of the long rny-lacZ fusion by RNase Y quite accurately mimics autoregulation of the wild-type rny gene. Intermediate length rny-lacZ fusions containing 13 and 31 rny codons, respectively, showed that with increasing length regulation becomes more efficient. The 31 codon fusion was regulated to the same extent as the long fusion suggesting that cleavages within the first 100 nucleotides of the rny ORF are important for control.
3.3. RNase Y Affects the Stability of Its Own mRNA
As shown above, it is the endonucleolytic activity of RNase Y which is required for the observed auto-control of rny expression. We therefore investigated whether changes in rny transcript levels involve modulation of rny mRNA stability. The half-life of the chromosomal rny transcript was measured in presence of physiological levels of RNase Y, i.e., B. subtilis cells containing the empty control plasmid (pC), or when the nuclease was overproduced (pY). Overproduction of RNase Y from pY is induced with tetracycline but since the high amount of the nuclease expressed under these conditions lead to undetectable rny mRNA levels (confer Figure 2A) we grew cells without inducer. As already mentioned, this still leads to ~two-fold excess of RNase Y in the cell. This overproduction of RNase Y caused a 3.5-fold decrease in rny half-life (1.2 min vs. 4 min) as shown in Figure 3A. We observed an equivalent four-fold effect when measuring the half-life of the long rny-lacZ fusion (Figure 3B), but a smaller two-fold reduction in half-life for the short rny-lacZ fusion (Figure 3C). This suggested that cleavage(s) in the 5′ proximal sequences of the rny ORF make the most significant contribution to auto-control.
3.4. RNase Y Can Cleave Efficiently in the 5′ UTR of its mRNA
The half-life and the intracellular level of the short rny-lacZ mRNA was still affected in the ∆rny strain or by increased levels of RNase Y, albeit to a lesser extent than the long fusion mRNA. This implied that RNase Y might not only cleave within the rny open reading frame but also in the 5′ UTR. In order to identify such cleavages, we analyzed the short fusion mRNA by primer extension in strains containing the inducible RNase Y overexpression plasmid pHMD22 (pY) grown with or without inducer or the control plasmid. The amount of full-length cDNA (238 nt) was reduced in the RNase Y overexpressing strain, especially when grown in the presence of inducer (Figure 4A). Under these conditions, a ~90 nt cDNA accumulated as would be expected if RNase Y cleavage products were trimmed by RNase J1 to the point when the 5′ exonuclease encounters a ribosome bound to the translation initiation site. Since the ribosome protects about −15 to +15 nt on the mRNA with respect to the initiation codon [51], a 5′-end at position −15 would result in a cDNA of 88 nt which is in line with the detected ~90 nt cDNA product.
In order to find out where RNase Y might cleave within the 5′ UTR, we carried out the primer extension analysis in an RNase J1 deletion mutant where the 5′ ends of cleavage products should be stabilized. Surprisingly, we found a large number of cleavages all along the 164 nt 5′ UTR (Figure 4B). In accordance, the multiple new 5′ ends detected within the 5′ UTR are almost absent in the wild-type strain expressing RNase J1. Instead, the major 5′ end identified within the 5′ UTR of the rny mRNA likely results from trimming by RNase J1 up to the position of a ribosome bound at the Shine–Dalgarno sequence as described above (Figure 4B).
Deletion of almost the entire 5′ UTR, residues −21 to −158 of the 164 nt leader, in the long rny-lacZ construct still allowed auto-control when overexpressing RNase Y, albeit at some reduced level as judged by Northern analysis (Figure 4C, left panel). However, in the absence of the 5′ UTR the short rny-lacZ fusion mRNA level was completely insensitive to RNase Y overexpression (Figure 4C, right panel), as would be expected from the absence of both regions we had identified as being sensitive to cleavage by RNase Y.
4. Discussion
In this study, we have shown that B. subtilis RNase Y can regulate its expression by cleaving its own mRNA. The use of rny-lacZ translational fusions was helpful as notably the long fusion encoding the 210 N-terminal amino acids of RNase Y behaved very similar when compared to the wild-type rny transcript. Auto-control is very sensitive, a 1.5-fold overexpression of RNase Y is sufficient to strongly reduce rny mRNA levels and the same rny transcripts are strongly increased in a strain lacking RNase Y.
This regulation is most likely direct and strictly requires the endonucleolytic activity of the enzyme. Even a strong overexpression of a catalytic mutant of RNase Y does not downregulate rny expression. Interestingly, rny transcript levels or rny-lacZ fusion activity are not only not reduced under these conditions but strongly induced. This is exactly the phenotype we observed in a ∆rny strain and implies that inactive RNase Y has a dominant negative effect on global RNase Y activity. Soluble forms of RNase Y lacking the N-terminal transmembrane domain are active in vitro [2] and form mainly dimers and tetramers [49]. The N-terminal non-catalytic domain of RNase Y which is intrinsically disordered contributes significantly to dimerization [50]. In vivo, RNase Y also forms dynamic higher order complexes whose assembly status can be shifted by regulatory proteins, with smaller complexes likely being the most active form of the enzyme [27]. Since RNase Y occurs, at a minimum, as a dimer, overproduction of inactive RNase Y proteins should lead to mixed forms of RNase Y oligomers where the mutated entity might also inhibit the activity of the wild-type form present in the same complex. Alternatively, but not mutually exclusive, mutated RNase Y might simply compete with the wild-type enzyme for binding to the rny mRNA and other substrates, imitating an apparent lack of RNase Y in the cell.
The use of translational rny-lacZ fusions proved to be very useful for characterizing the regions of the rny transcript susceptible to RNase Y cleavage. Early studies in B. subtilis on the role of stabilizing elements like secondary structure or a ribosome binding site at the 5′ end of an mRNA had shown that the E. coli lacZ mRNA fused to these upstream elements could actually be stabilized to attain half-lives exceeding 40 min [52,53]. This suggested that in B. subtilis the heterologous lacZ mRNA is quite resistant to endonucleolytic cleavage, notably by RNase Y, an enzyme unknown at the time. Here, we could clearly show that even a strong overexpression of RNase Y cannot reduce the intracellular level of a rny-lacZ fusion mRNA when those rny regions that are susceptible to RNase Y cleavage are replaced or deleted (Figure 4C).
RNase Y can cleave its mRNA at multiple positions to control the steady-state level of the functional transcript. Cleavages within the open reading frame would inactivate the transcript for a new translational round. We tried to identify specific cleavage sites within the rny ORF by a primer extension analysis of rny-lacZ fusions of various length in an RNase J1 mutant strain and obtained multiple but not well reproducible signals. While the long fusion containing 600 nt of rny sequences was the most susceptible to RNase Y cleavage, a significant effect of the absence of RNase Y on the level of rny-lacZ fusion mRNA was already observed with a fusion containing only 39 nt (13 codons) of rny. The presence of 93 nt (31 codons) of rny gave results equivalent to those of the long fusion. This indicates that RNase Y controls its own expression to a large part by cleaving within the first 100 nucleotides of the rny ORF. This might involve multiple cleavages as the absence of a 5′ exonucleolytic activity (RNase J1 mutant) was not enough to precisely identify new 5′ ends within the ORF. However, internal fragments created by more than one cleavage would also be degraded by 3′ exoribonucleases complicating their detection. Translation of the rny ORF does not appear to play an important role in regulation. A long lacZ fusion construct with an AUC start codon mutation was downregulated when RNase Y was overexpressed, similarly to the wild-type fusion albeit at much lower expression levels.
A second more indirect control involves RNase Y cleavages within the rny 5′ UTR. The many new 5′ ends detected in the RNase J1 mutant may not all be genuine RNase Y cleavage sites. However, overexpression of RNase Y clearly destabilizes the 5′ UTR producing entry sites for the 5′ exonuclease J1 which trims the mRNA until encountering an initiating ribosome. It remains a surprising finding that a 3 kb mRNA molecule like the lacZ transcript appears completely resistant to cleavage by RNase Y while the 160 nt rny 5′ UTR is cleaved efficiently at multiple positions. RNase Y is thought to cleave with little sequence specificity in single-stranded AU-rich sequences with a secondary structure in the up- or downstream vicinity and with a certain preference for cleavage at or close to a G residue [2,11,12,14]. We have observed that the strongest cleavages in the rny 5′ UTR occur within the fifty 5′ proximal nucleotides, a region composed of 76% U and A residues with a single stem-loop in the middle part. The 5′ proximal sequences of the rny ORF also contain several long A stretches interrupted by a few G or C residues. However, as expected we found no clearly defined characteristics that would allow a valid prediction of potential RNase Y cleavage sites, or explain why the lacZ mRNA is not a substrate.
The β-gal activity of the rny-lacZ fusions was generally in line with the transcript levels determined by Northern blot except in one case. Induced overexpression of RNase Y caused only a minor 1.3-fold effect on the β-gal activity of the short fusion reporter while the same conditions led to a 4-fold reduction in the levels of the short fusion mRNA (Figure 2B, confer lanes 1 to 4). This apparent difference might be linked to the length of the rny-lacZ transcript (3.2 kb). Assuming an average elongation rate of ~40 nucleotides/s, it should take about 80 s to synthesize the 3.2 kb transcript. Notably under conditions of RNase Y overexpression, most nascent rny-lacZ transcripts should begin to decay before their synthesis is completed. With the short rny-lacZ fusion, RNase Y cannot directly inactivate the transcript because the rny sequences within the ORF susceptible to cleavage are absent. However, efficient cleavage within the rny 5′ UTR by RNase Y still occurs. In this case, inactivation of the transcript relies on the progression of the incoming 5′ exonuclease RNase J1 which can be hampered by a bound ribosome, notably at the Shine–Dalgarno sequence. In this configuration, the translation of most rny-lacZ mRNAs can probably proceed to the end abidingly, even though the levels of the full-length transcripts are constantly decreased due to shortening of the mRNA from the 5′ end. Since actively translating transcripts would be less efficiently degraded from the 5′ end this could explain the discrepancy between β-gal activity and full-length short rny-lacZ mRNA levels. In accordance, the 5′ dependent degradation is relieved in short rny-lacZ transcripts when the 5′ UTR rny sequences that are susceptible to RNase Y cleavage are not present.
We have summarized the current data in a model for the regulation of expression of the rny gene (Figure 5A). It contrasts with that proposed for the autoregulation of RNase E in E. coli where binding of a stem-loop in the rne mRNA 5′ UTR, without being cleaved, seems required for expediting cleavage elsewhere within the rne transcript [54].
In this context, it is noteworthy that the rny Shine–Dalgarno sequence can form 10 canonical Watson–Crick and one G•U base pair with the 3′-end of 16S rRNA, with a ∆G of −17.5 kcal mol−1. This actually represents one of the strongest ribosomal binding sites present in B. subtilis mRNAs [56]. A strong interaction between the ribosome and the Shine–Dalgarno sequence might be necessary for the ribosome to efficiently block RNase J1 progression, but also to provide for efficient translation initiation especially since the RBS can be part of a strong secondary structure that extends up to codon 13 of the rny ORF (Figure 5B). Similar structures are also found in the rny transcripts of closely related species. Indeed, the secondary structure itself may also decelerate RNase J1 progression and such a possibility would be compatible with the observed ~90 nt cDNA product from the primer extension experiment. On the other hand, formation of the stem-loop structure could also play a role for the susceptibility of the translated mRNA to RNase Y cleavages occurring in the 5′ proximal coding sequences. Mutations predicted to destabilize the structure without altering the RBS had no significant effect on expression but slightly reduced the capacity of overexpressed RNase Y to downregulate expression. The protection of the rny transcript from 5′ exonucleolytic degradation by ribosome binding provides a sensitive link between translation initiation and the functional stability of the RNA. A number of aspects of this regulation need to be addressed in the future, notably the role of secondary structure formation at the translation initiation site and whether other factors potentially interacting with RNase Y can interfere with autoregulation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms11061374/s1. Original uncropped Western and Northern blots used for creating the figures in the manuscript.
Author Contributions
Conceptualization, S.L. and H.P.; methodology, A.K. and S.L.; validation, A.K., S.L. and H.P.; formal analysis, S.L. and H.P.; investigation, A.K. and C.B.; resources, H.P.; data curation, A.K. and H.P.; writing—original draft preparation, A.K.; writing—review and editing, H.P.; supervision, S.L. and H.P.; project administration, H.P.; funding acquisition, H.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Centre National de la Recherche Scientifique [UMR 8261], University Paris Cité, Agence Nationale de la Recherche [IB-mRND] and Labex Dynamo ANR-11-LABX-0011-01.
Data Availability Statement
Data sharing is not applicable for this article as no datasets were generated or analyzed during the current study.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Laalami, S.; Zig, L.; Putzer, H. Initiation of mRNA decay in bacteria. Cell. Mol. Life Sci. 2014, 71, 1799–1828. [Google Scholar] [CrossRef] [PubMed]
- Shahbabian, K.; Jamalli, A.; Zig, L.; Putzer, H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009, 28, 3523–3533. [Google Scholar] [CrossRef]
- Lehnik-Habrink, M.; Schaffer, M.; Mader, U.; Diethmaier, C.; Herzberg, C.; Stulke, J. RNA processing in Bacillus subtilis: Identification of targets of the essential RNase Y. Mol. Microbiol. 2011, 81, 1459–1473. [Google Scholar] [CrossRef]
- Durand, S.; Gilet, L.; Bessieres, P.; Nicolas, P.; Condon, C. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet. 2012, 8, e1002520. [Google Scholar] [CrossRef] [PubMed]
- Laalami, S.; Bessieres, P.; Rocca, A.; Zig, L.; Nicolas, P.; Putzer, H. Bacillus subtilis RNase Y activity in vivo analysed by tiling microarrays. PLoS ONE 2013, 8, e54062. [Google Scholar] [CrossRef]
- Richards, J.; Liu, Q.; Pellegrini, O.; Celesnik, H.; Yao, S.; Bechhofer, D.H.; Condon, C.; Belasco, J.G. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol. Cell 2011, 43, 940–949. [Google Scholar] [CrossRef]
- Even, S.; Pellegrini, O.; Zig, L.; Labas, V.; Vinh, J.; Brechemmier-Baey, D.; Putzer, H. Ribonucleases J1 and J2: Two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 2005, 33, 2141–2152. [Google Scholar] [CrossRef]
- Li de la Sierra-Gallay, I.; Zig, L.; Jamalli, A.; Putzer, H. Structural insights into the dual activity of RNase J. Nat. Struct. Mol. Biol. 2008, 15, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Mathy, N.; Benard, L.; Pellegrini, O.; Daou, R.; Wen, T.; Condon, C. 5′-to-3′ Exoribonuclease Activity in Bacteria: Role of RNase J1 in rRNA Maturation and 5′ Stability of mRNA. Cell 2007, 129, 681–692. [Google Scholar] [CrossRef]
- Mathy, N.; Hebert, A.; Mervelet, P.; Benard, L.; Dorleans, A.; Li de la Sierra-Gallay, I.; Noirot, P.; Putzer, H.; Condon, C. Bacillus subtilis ribonucleases J1 and J2 form a complex with altered enzyme behaviour. Mol. Microbiol. 2010, 75, 489–498. [Google Scholar] [CrossRef]
- Khemici, V.; Prados, J.; Linder, P.; Redder, P. Decay-Initiating Endoribonucleolytic Cleavage by RNase Y Is Kept under Tight Control via Sequence Preference and Sub-cellular Localisation. PLoS Genet. 2015, 11, e1005577. [Google Scholar] [CrossRef]
- Marincola, G.; Wolz, C. Downstream element determines RNase Y cleavage of the saePQRS operon in Staphylococcus aureus. Nucleic Acids Res. 2017, 45, 5980–5994. [Google Scholar] [CrossRef]
- DeLoughery, A.; Lalanne, J.B.; Losick, R.; Li, G.W. Maturation of polycistronic mRNAs by the endoribonuclease RNase Y and its associated Y-complex in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2018, 115, E5585–E5594. [Google Scholar] [CrossRef] [PubMed]
- Broglia, L.; Lecrivain, A.L.; Renault, T.T.; Hahnke, K.; Ahmed-Begrich, R.; Le Rhun, A.; Charpentier, E. An RNA-seq based comparative approach reveals the transcriptome-wide interplay between 3′-to-5′ exoRNases and RNase Y. Nat. Commun. 2020, 11, 1587. [Google Scholar] [CrossRef] [PubMed]
- Ehretsmann, C.; Carpousis, A.J.; Krisch, H.M. Specificity of Escherichia coli endoribonuclease RNAse E: In vivo and in vitro analysis of mutants in a bacteriophage T4 mRNA processing site. Genes Dev. 1992, 6, 149–159. [Google Scholar] [CrossRef]
- Mackie, G.A. Secondary structure of the mRNA for ribosomal protein S20. Implications for cleavage by ribonuclease E. J. Biol. Chem. 1992, 267, 1054–1061. [Google Scholar] [CrossRef]
- McDowall, K.J.; Lin-Chao, S.; Cohen, S.N. A + U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J. Biol. Chem. 1994, 269, 10790–10796. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Li, L.; Girodat, D.; Forstner, K.U.; Said, N.; Corcoran, C.; Smiga, M.; Papenfort, K.; Reinhardt, R.; Wieden, H.J.; et al. In Vivo Cleavage Map Illuminates the Central Role of RNase E in Coding and Non-coding RNA Pathways. Mol. Cell 2017, 65, 39–51. [Google Scholar] [CrossRef]
- Bandyra, K.J.; Luisi, B.F. RNase E and the High-Fidelity Orchestration of RNA Metabolism. Microbiol. Spectr. 2018, 6, 23. [Google Scholar] [CrossRef]
- Del Campo, C.; Bartholomaus, A.; Fedyunin, I.; Ignatova, Z. Secondary Structure across the Bacterial Transcriptome Reveals Versatile Roles in mRNA Regulation and Function. PLoS Genet. 2015, 11, e1005613. [Google Scholar] [CrossRef]
- Laalami, S.; Cavaiuolo, M.; Roque, S.; Chagneau, C.; Putzer, H. Escherichia coli RNase E can efficiently replace RNase Y in Bacillus subtilis. Nucleic Acids Res. 2021, 49, 4643–4654. [Google Scholar] [CrossRef]
- Khemici, V.; Poljak, L.; Luisi, B.F.; Carpousis, A.J. The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 2008, 70, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Strahl, H.; Turlan, C.; Khalid, S.; Bond, P.J.; Kebalo, J.M.; Peyron, P.; Poljak, L.; Bouvier, M.; Hamoen, L.; Luisi, B.F.; et al. Membrane recognition and dynamics of the RNA degradosome. PLoS Genet. 2015, 11, e1004961. [Google Scholar] [CrossRef]
- Hunt, A.; Rawlins, J.P.; Thomaides, H.B.; Errington, J. Functional analysis of 11 putative essential genes in Bacillus subtilis. Microbiology 2006, 152, 2895–2907. [Google Scholar] [CrossRef]
- Lewis, P.J.; Thaker, S.D.; Errington, J. Compartmentalization of transcription and translation in Bacillus subtilis. EMBO J. 2000, 19, 710–718. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Weber, M.H.; Graumann, P.L. Specific polar localization of ribosomes in Bacillus subtilis depends on active transcription. EMBO Rep. 2001, 2, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Hamouche, L.; Billaudeau, C.; Rocca, A.; Chastanet, A.; Ngo, S.; Laalami, S.; Putzer, H. Dynamic Membrane Localization of RNase Y in Bacillus subtilis. mBio 2020, 11, e03337-19. [Google Scholar] [CrossRef]
- Commichau, F.M.; Rothe, F.M.; Herzberg, C.; Wagner, E.; Hellwig, D.; Lehnik-Habrink, M.; Hammer, E.; Völker, U.; Stülke, J. Novel activities of glycolytic enzymes in Bacillus subtilis: Interactions with essential proteins involved in mRNA processing. Mol. Cell Proteom. 2009, 8, 1350–1360. [Google Scholar] [CrossRef]
- Lehnik-Habrink, M.; Pfortner, H.; Rempeters, L.; Pietack, N.; Herzberg, C.; Stülke, J. The RNA degradosome in Bacillus subtilis: Identification of CshA as the major RNA helicase in the multiprotein complex. Mol. Microbiol. 2010, 77, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Carpousis, A.J.; Van Houwe, G.; Ehretsmann, C.; Krisch, H.M. Copurification of E. coli RNAse E and PNPase: Evidence for a specific association between two enzymes important in mRNA processing and degradaion. Cell 1994, 76, 889–900. [Google Scholar] [CrossRef]
- Gao, J.; Lee, K.; Zhao, M.; Qiu, J.; Zhan, X.; Saxena, A.; Moore, C.J.; Cohen, S.N.; Georgiou, G. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol. Microbiol. 2006, 61, 394–406. [Google Scholar] [CrossRef]
- Cascante-Estepa, N.; Gunka, K.; Stulke, J. Localization of Components of the RNA-Degrading Machine in Bacillus subtilis. Front. Microbiol. 2016, 7, 1492. [Google Scholar] [CrossRef]
- Newman, J.A.; Hewitt, L.; Rodrigues, C.; Solovyova, A.S.; Harwood, C.R.; Lewis, R.J. Dissection of the network of interactions that links RNA processing with glycolysis in the Bacillus subtilis degradosome. J. Mol. Biol. 2012, 416, 121–136. [Google Scholar] [CrossRef]
- Redder, P. Molecular and genetic interactions of the RNA degradation machineries in Firmicute bacteria. Wiley Interdiscip. Rev. RNA 2018, 9, e1460. [Google Scholar] [CrossRef]
- Adusei-Danso, F.; Khaja, F.T.; DeSantis, M.; Jeffrey, P.D.; Dubnau, E.; Demeler, B.; Neiditch, M.B.; Dubnau, D. Structure-Function Studies of the Bacillus subtilis Ric Proteins Identify the Fe-S Cluster-Ligating Residues and Their Roles in Development and RNA Processing. mBio 2019, 10, e01841-19. [Google Scholar] [CrossRef]
- Carabetta, V.J.; Tanner, A.W.; Greco, T.M.; Defrancesco, M.; Cristea, I.M.; Dubnau, D. A complex of YlbF, YmcA and YaaT regulates sporulation, competence and biofilm formation by accelerating the phosphorylation of Spo0A. Mol. Microbiol. 2013, 88, 283–300. [Google Scholar] [CrossRef]
- DeLoughery, A.; Dengler, V.; Chai, Y.; Losick, R. Biofilm formation by Bacillus subtilis requires an endoribonuclease-containing multisubunit complex that controls mRNA levels for the matrix gene repressor SinR. Mol. Microbiol. 2016, 99, 425–437. [Google Scholar] [CrossRef]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Messing, J. New M13 vectors for cloning genes. In Methods in Enzymology; Wu, R., Grossman, L., Moldave, K., Eds.; Academic Press: New York, NY, USA, 1983; Volume 101, Part C, pp. 20–89. [Google Scholar]
- Jamalli, A.; Hebert, A.; Zig, L.; Putzer, H. Control of expression of the RNases J1 and J2 in Bacillus subtilis. J. Bacteriol. 2014, 196, 318–324. [Google Scholar] [CrossRef]
- Guerout-Fleury, A.M.; Shazand, K.; Frandsen, N.; Stragier, P. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 1995, 167, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Heusterspreute, M.; Thi, V.H. Vectors with restriction site banks. IV. pJRD184, a 3793-bp plasmid vector having 43 unique cloning sites. Gene 1985, 39, 299–304. [Google Scholar] [CrossRef]
- Geissendorfer, M.; Hillen, W. Regulated expression of heterologous genes in Bacillus subtilis using the Tn10 encoded tet regulatory elements. Appl. Microbiol. Biotechnol. 1990, 33, 657–663. [Google Scholar] [CrossRef]
- Guerout-Fleury, A.M.; Frandsen, N.; Stragier, P. Plasmids for ectopic integration in Bacillus subtilis. Gene 1996, 180, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Putzer, H.; Brackhage, A.A.; Grunberg-Manago, M. Independent genes for two threonyl-tRNA synthetases in Bacillus subtilis. J. Bacteriol. 1990, 172, 4593–4602. [Google Scholar] [CrossRef]
- Putzer, H.; Gendron, N.; Grunberg-Manago, M. Co-ordinate expression of the two threonyl-tRNA synthetase genes in Bacillus subtilis: Control by transcriptional antitermination involving a conserved regulatory sequence. EMBO J. 1992, 11, 3117–3127. [Google Scholar] [CrossRef]
- Irnov, I.; Sharma, C.M.; Vogel, J.; Winkler, W.C. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010, 38, 6637–6651. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Lehnik-Habrink, M.; Newman, J.; Rothe, F.M.; Solovyova, A.S.; Rodrigues, C.; Herzberg, C.; Commichau, F.M.; Lewis, R.J.; Stulke, J. RNase Y in Bacillus subtilis: A Natively disordered protein that is the functional equivalent of RNase E from Escherichia coli. J. Bacteriol. 2011, 193, 5431–5441. [Google Scholar] [CrossRef] [PubMed]
- Hardouin, P.; Velours, C.; Bou-Nader, C.; Assrir, N.; Laalami, S.; Putzer, H.; Durand, D.; Golinelli-Pimpaneau, B. Dissociation of the Dimer of the Intrinsically Disordered Domain of RNase Y upon Antibody Binding. Biophys. J. 2018, 115, 2102–2113. [Google Scholar] [CrossRef]
- Yusupova, G.Z.; Yusupov, M.M.; Cate, J.H.; Noller, H.F. The path of messenger RNA through the ribosome. Cell 2001, 106, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hue, K.K.; Cohen, S.D.; Bechhofer, D.H. A polypurine sequence that acts as a 5′ mRNA stabilizer in Bacillus subtilis. J. Bacteriol. 1995, 177, 3465–3471. [Google Scholar] [CrossRef]
- Hambraeus, G.; Karhumaa, K.; Rutberg, B. A 5′ stem-loop and ribosome binding but not translation are important for the stability of Bacillus subtilis aprE leader mRNA. Microbiology 2002, 148, 1795–1803. [Google Scholar] [CrossRef]
- Schuck, A.; Diwa, A.; Belasco, J.G. RNase E autoregulates its synthesis in Escherichia coli by binding directly to a stem-loop in the rne 5′ untranslated region. Mol. Microbiol. 2009, 72, 470–478. [Google Scholar] [CrossRef]
- Vienna RNA Webservers. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi (accessed on 10 May 2023).
- Wei, Y.; Silke, J.R.; Xia, X. Elucidating the 16S rRNA 3′ boundaries and defining optimal SD/aSD pairing in Escherichia coli and Bacillus subtilis using RNA-Seq data. Sci. Rep. 2017, 7, 17639. [Google Scholar] [CrossRef]
Figure 1.
Effect of RNase Y on the activity of rny-lacZ fusions. (A). Domains composing B. subtilis RNase Y (520 aa) include an N-terminal transmembrane domain (TMD, aa 1–25), followed by a large region predicted to be disordered (aa~30–210), an RNA binding KH domain (aa 211–270), and a metal-chelating HD domain (aa 336–429) containing the conserved His/Asp motif required for RNase activity [2,24,48,49,50]. Ab indicates the position of the 12 aa peptide used for monoclonal antibody production. (B). Sequence of the sigma A-type promoter of the rny gene and schematic of the «short» and «long» translational rny-lacZ fusions; the −10 and −35 regions and the transcription start site (+1) are indicated. (C). β-galactosidase activity of short and long rny-lacZ fusions in strains containing either the empty control plasmid (pC) or plasmids expressing wild-type RNase Y (pY) or an inactive version of RNase Y (pY*) in the presence of inducer (0.2 mg/mL of tetracycline). All cultures were grown in the presence of inducer and the amount of RNase Y in the cell was determined by Western blot using an RNase Y-specific antibody (lower panels). The antibody also detected the long lacZ fusion protein. (D). β-galactosidase activity of long and short rny-lacZ fusions in the wild-type and rny deletion (∆Y) strain. RNase Y expression and the absence of RNase Y in the mutant strain was verified by Western blot (lower panel). Y = RNase Y. Numbers below the Western blots show relative densitometry readings (Dens) based on the control value arbitrarily set to 100 in each gel. Measurements refer to the bands corresponding to RNase Y, except in (D) where they correspond to the fusion protein.
Figure 1.
Effect of RNase Y on the activity of rny-lacZ fusions. (A). Domains composing B. subtilis RNase Y (520 aa) include an N-terminal transmembrane domain (TMD, aa 1–25), followed by a large region predicted to be disordered (aa~30–210), an RNA binding KH domain (aa 211–270), and a metal-chelating HD domain (aa 336–429) containing the conserved His/Asp motif required for RNase activity [2,24,48,49,50]. Ab indicates the position of the 12 aa peptide used for monoclonal antibody production. (B). Sequence of the sigma A-type promoter of the rny gene and schematic of the «short» and «long» translational rny-lacZ fusions; the −10 and −35 regions and the transcription start site (+1) are indicated. (C). β-galactosidase activity of short and long rny-lacZ fusions in strains containing either the empty control plasmid (pC) or plasmids expressing wild-type RNase Y (pY) or an inactive version of RNase Y (pY*) in the presence of inducer (0.2 mg/mL of tetracycline). All cultures were grown in the presence of inducer and the amount of RNase Y in the cell was determined by Western blot using an RNase Y-specific antibody (lower panels). The antibody also detected the long lacZ fusion protein. (D). β-galactosidase activity of long and short rny-lacZ fusions in the wild-type and rny deletion (∆Y) strain. RNase Y expression and the absence of RNase Y in the mutant strain was verified by Western blot (lower panel). Y = RNase Y. Numbers below the Western blots show relative densitometry readings (Dens) based on the control value arbitrarily set to 100 in each gel. Measurements refer to the bands corresponding to RNase Y, except in (D) where they correspond to the fusion protein.
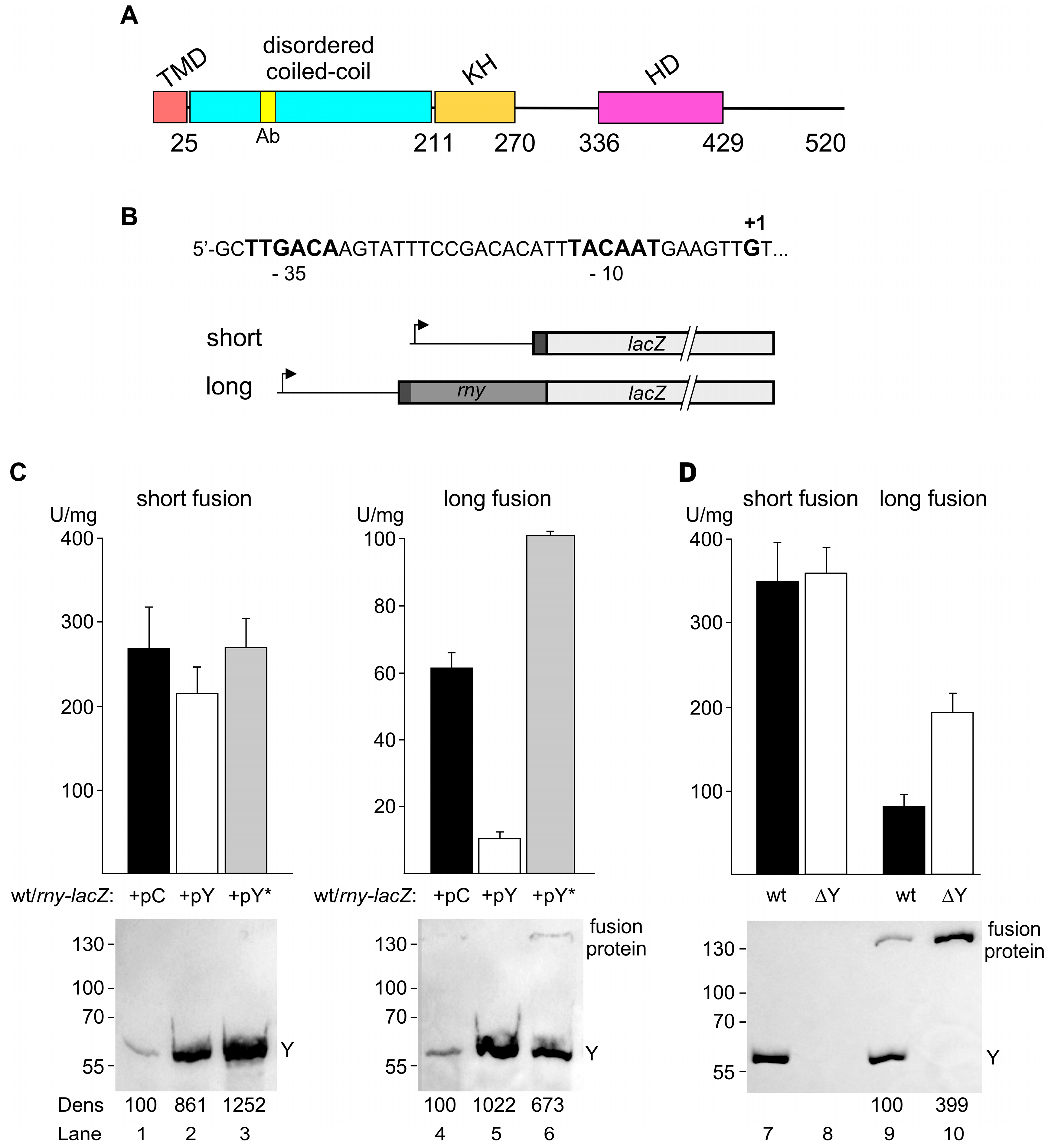
Figure 2.
Effect of RNase Y on wild-type rny and rny-lacZ transcript levels. (A). Northern blot analysis of rny mRNA (1.72 kb) in the wild-type (SSB1002) and strains containing either the control plasmid (pC, SSB614) or plasmids conditionally overexpressing wild-type RNase Y (pY, SSB615) or an inactive version of RNase Y (pY*, SSB616). Cultures were grown with or without inducer (0.2 μg/mL tetracycline) as indicated. RNase Y levels were determined by Western blot (lower panel) (B). Northern blot analysis of short and long rny-lacZ transcripts (3.2 and 3.8 kb, respectively) in strains containing either the control (pC, SSB1082 and SSB1084) or the RNase Y conditionally overexpressing plasmid (pY, SSB1083 and SSB1085). Transcripts were detected using a lacZ-specific probe. Cultures were grown with or without inducer (0.2 μg/mL tetracycline) as indicated. RNase Y levels were determined by Western blot (lower panel) (C). Northern blot analysis of rny-lacZ transcripts in wild-type and a Δrny background using a lacZ-specific probe. Wild-type strains (lanes 1, 3, 5, 7): SSB525, SSB530, SSB599, SSB600. Delta-rny strains (lanes 2, 4, 6, 8): SSB593, SSB594, SSB602, SSB603. All cultures were grown in LB medium. Hybridization with a 16S rRNA specific probe was used as a loading control. Numbers below the Northern and Western blots show relative densitometry readings based on the control value arbitrarily set to 100 in each gel. Measurements for Northern blots are already corrected for the 16S loading controls. NB: Northern blot. WB: Western blot.
Figure 2.
Effect of RNase Y on wild-type rny and rny-lacZ transcript levels. (A). Northern blot analysis of rny mRNA (1.72 kb) in the wild-type (SSB1002) and strains containing either the control plasmid (pC, SSB614) or plasmids conditionally overexpressing wild-type RNase Y (pY, SSB615) or an inactive version of RNase Y (pY*, SSB616). Cultures were grown with or without inducer (0.2 μg/mL tetracycline) as indicated. RNase Y levels were determined by Western blot (lower panel) (B). Northern blot analysis of short and long rny-lacZ transcripts (3.2 and 3.8 kb, respectively) in strains containing either the control (pC, SSB1082 and SSB1084) or the RNase Y conditionally overexpressing plasmid (pY, SSB1083 and SSB1085). Transcripts were detected using a lacZ-specific probe. Cultures were grown with or without inducer (0.2 μg/mL tetracycline) as indicated. RNase Y levels were determined by Western blot (lower panel) (C). Northern blot analysis of rny-lacZ transcripts in wild-type and a Δrny background using a lacZ-specific probe. Wild-type strains (lanes 1, 3, 5, 7): SSB525, SSB530, SSB599, SSB600. Delta-rny strains (lanes 2, 4, 6, 8): SSB593, SSB594, SSB602, SSB603. All cultures were grown in LB medium. Hybridization with a 16S rRNA specific probe was used as a loading control. Numbers below the Northern and Western blots show relative densitometry readings based on the control value arbitrarily set to 100 in each gel. Measurements for Northern blots are already corrected for the 16S loading controls. NB: Northern blot. WB: Western blot.
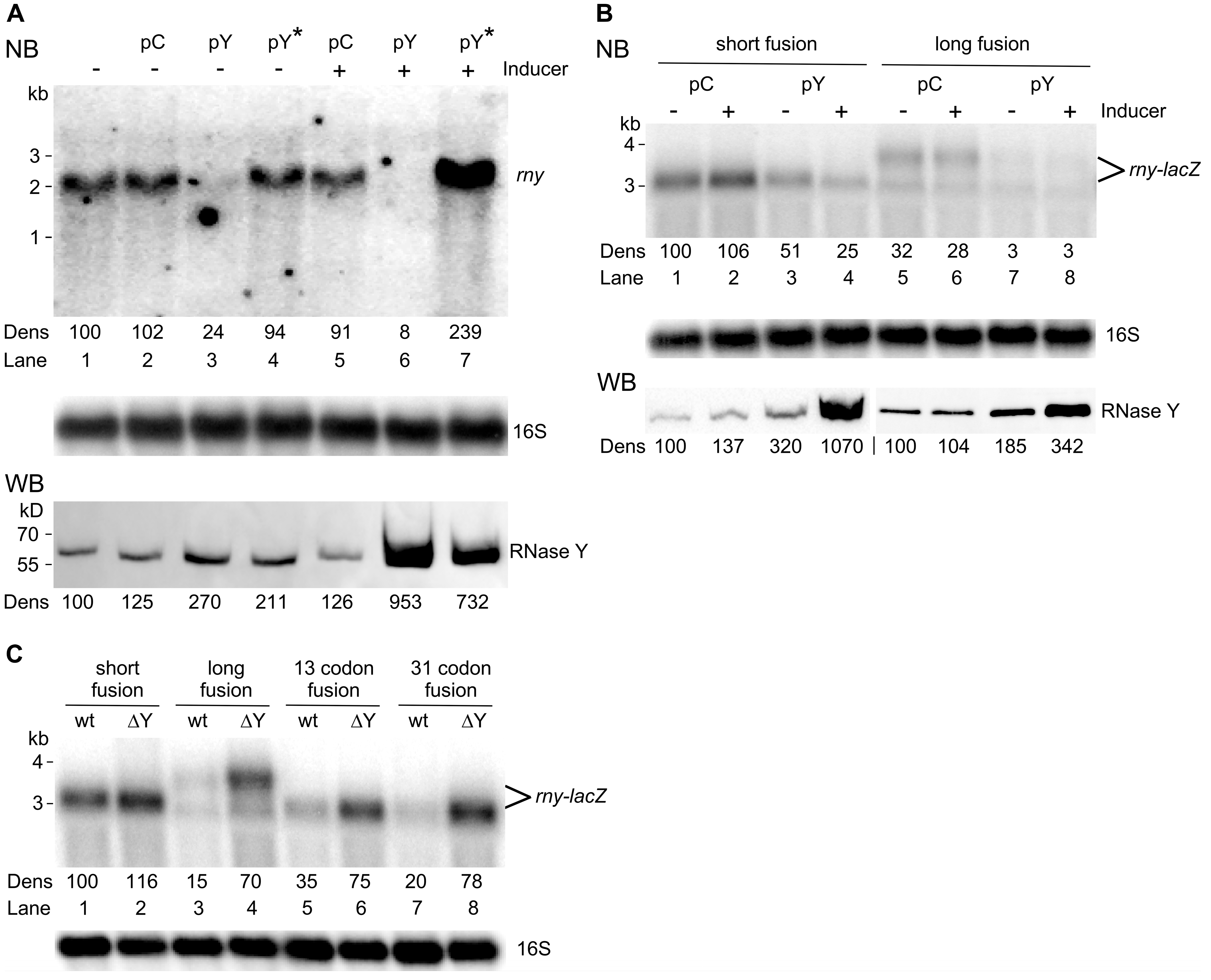
Figure 3.
Effect of RNase Y levels on the half-life of rny and rny-lacZ transcripts. (A). Half-life of wild-type rny mRNA in B. subtilis wild-type strains harboring either the empty vector (pC, SSB614) or the RNase Y overproducing plasmid (pY, SSB615). Rifampicin was added to mid-log cultures (time 0′) and RNA isolated at the indicated time points was hybridized to a probe specific for the rny 5′ UTR; this probe detects the chromosomally encoded rny mRNA and the rny-lacZ fusion mRNAs, but not the plasmid-encoded rny mRNA. The intracellular RNase Y level at time 0 was determined by Western blot (lower panels). The position of the rny transcript on the Northern blots is indicated. (B). Half-life measurement of the long rny-lacZ fusion mRNA in wild-type (SSB530) and ∆rny (SSB594) strains. Total RNA was isolated at the indicated time points and rny-lacZ transcripts were detected with a lacZ-specific probe. (C). Half-life measurement of the short rny-lacZ fusion mRNA in wild-type (SSB525) and ∆rny (SSB593) strains. Transcripts were detected with a lacZ-specific probe. All cultures were grown in LB medium and no inducer was added for RNase Y overexpression. Hybridization with a 16S rRNA specific probe was used as a loading control. Numbers below the Northern and Western blots (Dens) show relative densitometry readings based on the control value arbitrarily set to 100 in each gel. Measurements for Northern blots are already corrected for the 16S loading controls.
Figure 3.
Effect of RNase Y levels on the half-life of rny and rny-lacZ transcripts. (A). Half-life of wild-type rny mRNA in B. subtilis wild-type strains harboring either the empty vector (pC, SSB614) or the RNase Y overproducing plasmid (pY, SSB615). Rifampicin was added to mid-log cultures (time 0′) and RNA isolated at the indicated time points was hybridized to a probe specific for the rny 5′ UTR; this probe detects the chromosomally encoded rny mRNA and the rny-lacZ fusion mRNAs, but not the plasmid-encoded rny mRNA. The intracellular RNase Y level at time 0 was determined by Western blot (lower panels). The position of the rny transcript on the Northern blots is indicated. (B). Half-life measurement of the long rny-lacZ fusion mRNA in wild-type (SSB530) and ∆rny (SSB594) strains. Total RNA was isolated at the indicated time points and rny-lacZ transcripts were detected with a lacZ-specific probe. (C). Half-life measurement of the short rny-lacZ fusion mRNA in wild-type (SSB525) and ∆rny (SSB593) strains. Transcripts were detected with a lacZ-specific probe. All cultures were grown in LB medium and no inducer was added for RNase Y overexpression. Hybridization with a 16S rRNA specific probe was used as a loading control. Numbers below the Northern and Western blots (Dens) show relative densitometry readings based on the control value arbitrarily set to 100 in each gel. Measurements for Northern blots are already corrected for the 16S loading controls.
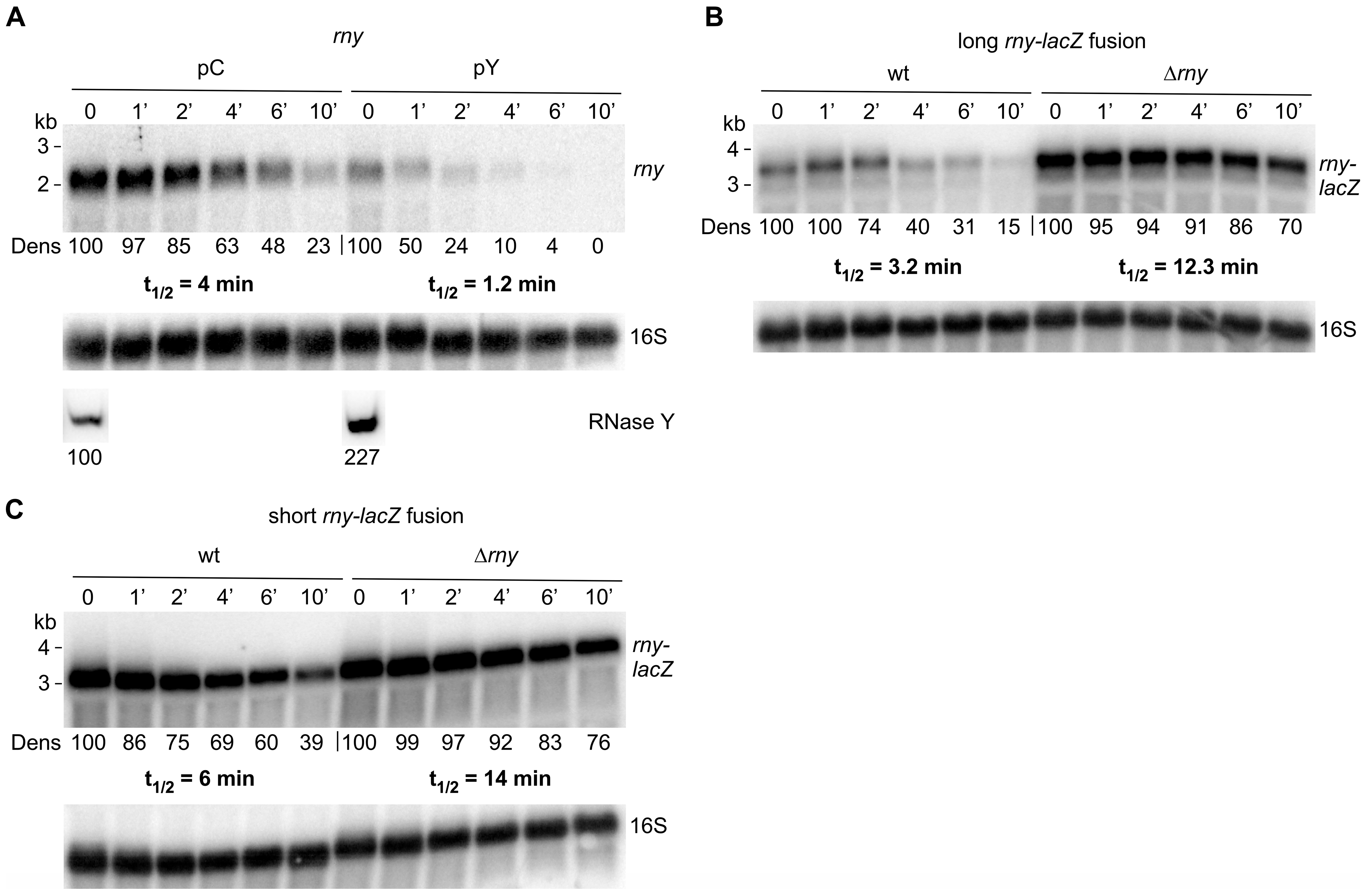
Figure 4.
RNase Y cleaves at multiple sites in the rny 5′ UTR. (A). Primer extension analysis of the short rny-lacZ fusion mRNA using a lacZ-specific primer (HP905, the 5′ end of the primer is complementary to lacZ sequences 57 nt downstream of the last rny codon) in strains harboring the empty control plasmid (pC, SSB1082) or the conditionally RNase Y overexpressing plasmid (pY, SSB1083). Cells were grown in the absence or presence of inducer (0.2 mg/mL of tetracycline). An arrow indicates the putative mRNA 5′ end generated by the 5′ exonuclease activity of RNase J1 and arrested by a ribosome bound to the Shine–Dalgarno sequence (15 nucleotides upstream of the AUG initiation codon corresponds to a cDNA of 88 nt). (B). Primer extension analysis of the short rny-lacZ fusion mRNA using a lacZ-specific primer (HP905) in a wild-type (SSB525) and RNase J1 mutant strain (∆J1, SSB1086). Extension of the primer to the original 5′ end leads to a product of 238 nt. The major 5′ end within the 5′ UTR likely generated by 5′ exonuclease trimming is indicated by an arrow. M: 25 bp ladder. (C). Effect of RNase Y overexpression on long and short rny-lacZ fusion transcripts that lack the rny 5′ UTR. Total RNA from strains carrying the long ∆5′ UTR rny-lacZ fusion and the empty control plasmid (pC, SSB638) or the RNase Y overexpression plasmid (pY, SSB639), and from strains carrying the short ∆5′ UTR rny-lacZ fusion and the empty control plasmid (pC, SSB630) or the RNase Y overexpression plasmid (pY, SSB631) was hybridized with a lacZ-specific probe. Numbers below the Northern blot show relative densitometry readings of the lacZ fusion mRNA based on the control value arbitrarily set to 100 in each gel. Measurements for Northern blots are already corrected for the 16S loading controls. Asterisks mark the position of ribosomal RNA hybridizing non-specifically to the lacZ probe.
Figure 4.
RNase Y cleaves at multiple sites in the rny 5′ UTR. (A). Primer extension analysis of the short rny-lacZ fusion mRNA using a lacZ-specific primer (HP905, the 5′ end of the primer is complementary to lacZ sequences 57 nt downstream of the last rny codon) in strains harboring the empty control plasmid (pC, SSB1082) or the conditionally RNase Y overexpressing plasmid (pY, SSB1083). Cells were grown in the absence or presence of inducer (0.2 mg/mL of tetracycline). An arrow indicates the putative mRNA 5′ end generated by the 5′ exonuclease activity of RNase J1 and arrested by a ribosome bound to the Shine–Dalgarno sequence (15 nucleotides upstream of the AUG initiation codon corresponds to a cDNA of 88 nt). (B). Primer extension analysis of the short rny-lacZ fusion mRNA using a lacZ-specific primer (HP905) in a wild-type (SSB525) and RNase J1 mutant strain (∆J1, SSB1086). Extension of the primer to the original 5′ end leads to a product of 238 nt. The major 5′ end within the 5′ UTR likely generated by 5′ exonuclease trimming is indicated by an arrow. M: 25 bp ladder. (C). Effect of RNase Y overexpression on long and short rny-lacZ fusion transcripts that lack the rny 5′ UTR. Total RNA from strains carrying the long ∆5′ UTR rny-lacZ fusion and the empty control plasmid (pC, SSB638) or the RNase Y overexpression plasmid (pY, SSB639), and from strains carrying the short ∆5′ UTR rny-lacZ fusion and the empty control plasmid (pC, SSB630) or the RNase Y overexpression plasmid (pY, SSB631) was hybridized with a lacZ-specific probe. Numbers below the Northern blot show relative densitometry readings of the lacZ fusion mRNA based on the control value arbitrarily set to 100 in each gel. Measurements for Northern blots are already corrected for the 16S loading controls. Asterisks mark the position of ribosomal RNA hybridizing non-specifically to the lacZ probe.
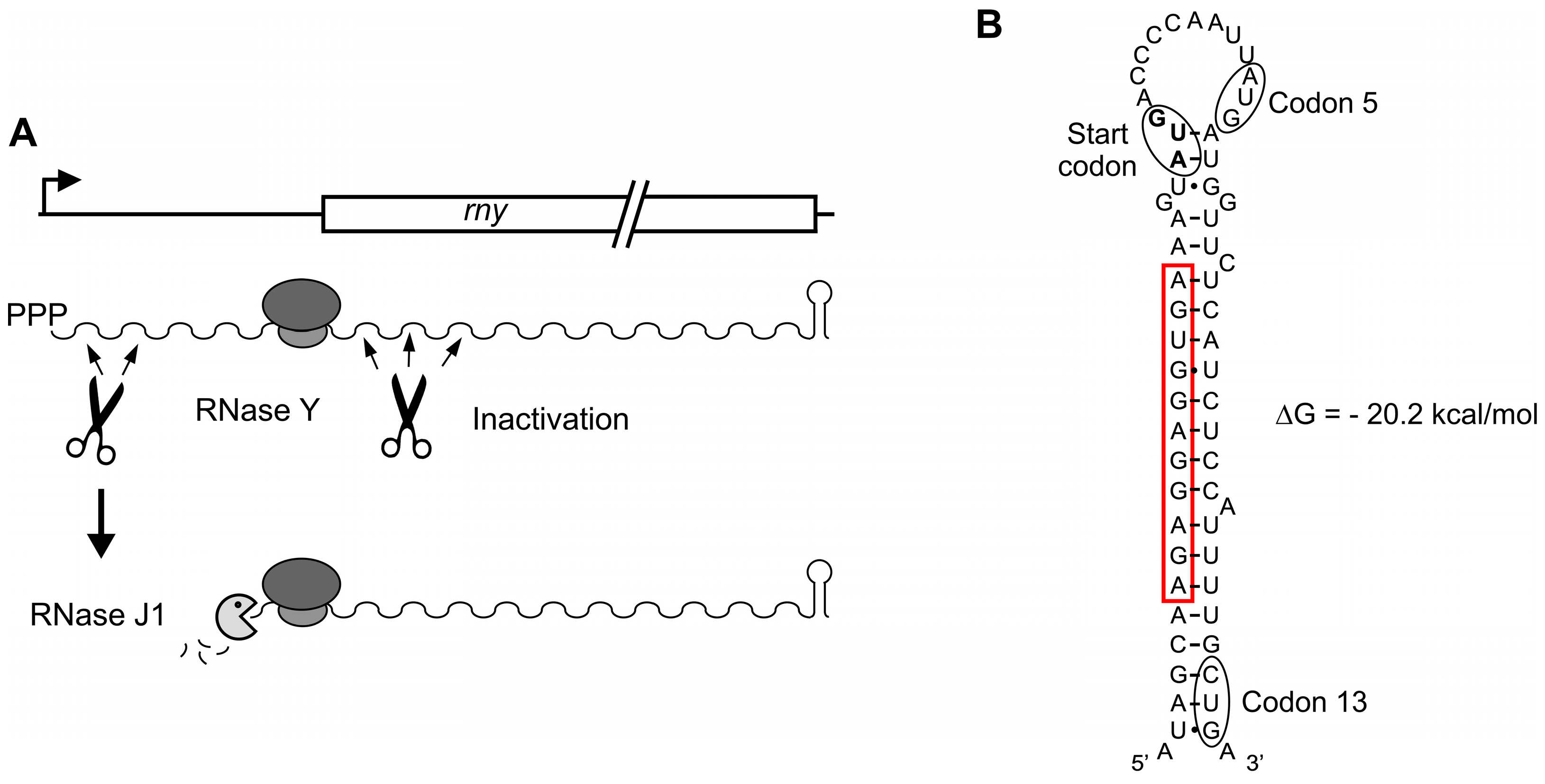
Figure 5.
Model of autoregulation of RNase Y synthesis. (A). Freely available RNase Y can downregulate its own synthesis by cleaving within the ORF and the 5′ UTR. Cleavage in the ORF likely at multiple positions immediately inactivates the transcript. On the contrary, cleavage within the 5′ UTR generates 5′ monophosphorylated entry sites for the 5′ exoribonuclease activity of RNase J1. In this case, inactivation of the rny transcript depends on ribosome occupancy at the translation initiation site, hence linking the degradation of the rny mRNA to translation efficiency. (B). Potential secondary structure of the translation initiation region predicted by RNAfold [55]. The Shine–Dalgarno sequence is boxed in red.
Figure 5.
Model of autoregulation of RNase Y synthesis. (A). Freely available RNase Y can downregulate its own synthesis by cleaving within the ORF and the 5′ UTR. Cleavage in the ORF likely at multiple positions immediately inactivates the transcript. On the contrary, cleavage within the 5′ UTR generates 5′ monophosphorylated entry sites for the 5′ exoribonuclease activity of RNase J1. In this case, inactivation of the rny transcript depends on ribosome occupancy at the translation initiation site, hence linking the degradation of the rny mRNA to translation efficiency. (B). Potential secondary structure of the translation initiation region predicted by RNAfold [55]. The Shine–Dalgarno sequence is boxed in red.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
B. subtilis strains used in this study.
| B. subtilis Strain | Relevant Genotype | Reference |
|---|---|---|
| SSB1002 | Wild-type strain | Lab stock |
| SSB525 | amyE::rny-lacZ (short, 5 aa) | This work |
| SSB530 | amyE::rny-lacZ (long, 210 aa) | This work |
| SSB593 | amyE::rny-lacZ (short, 5 aa), ∆rny::spc | This work |
| SSB594 | amyE::rny-lacZ (long, 210 aa), ∆rny::spc | This work |
| SSB599 | amyE::rny-lacZ (13 aa) | This work |
| SSB600 | amyE::rny-lacZ (31 aa) | This work |
| SSB602 | amyE::rny-lacZ (13 aa), ∆rny::spc | This work |
| SSB603 | amyE::rny-lacZ (31 aa), ∆rny::spc | This work |
| SSB614 | SSB1002, pWH353m | This work |
| SSB615 | SSB1002, pHMD22 | This work |
| SSB616 | SSB1002, pHMD33 | This work |
| SSB630 | amyE::rny-lacZ (short, 5 aa, ∆5′ UTR), pWH353 | This work |
| SSB631 | amyE::rny-lacZ (short, 5 aa, ∆5′ UTR), pHMD22 | This work |
| SSB638 | amyE::rny-lacZ (long, 210 aa, ∆5′ UTR), pWH353 | This work |
| SSB639 | amyE::rny-lacZ (long, 210 aa, ∆5′ UTR), pHMD22 | This work |
| SSB1082 | amyE::rny-lacZ (short, 5 aa), pWH353m | This work |
| SSB1083 | amyE::rny-lacZ (short, 5 aa), pHMD22 | This work |
| SSB1084 | amyE::rny-lacZ (long, 210 aa), pWH353m | This work |
| SSB1085 | amyE::rny-lacZ (long, 210 aa), pHMD22 | This work |
| SSB1086 | amyE::rny-lacZ (short, 5 aa), ∆rnjA::tet | This work |
“Short, 5aa” and “Long, 210 aa” refers to the number of rny codons present in the short and long rny-lacZ fusions, respectively.
Table 2.
Oligonucleotides used in this study.
| Oligonucleotide | Sequence 5′–3′ |
|---|---|
| HP899 | ACTGAGAATTCACGGTTTTTCTCGTACTTTCCGGT |
| HP905 | GATTAAGTTGGGTAACGCCAGGGT |
| HP1024 | ATTACCTGCAGACCGCTCATAACTCCAATAC |
| HP1039 | ACTATCTCGAGTCCTGCCGATCATTATGGAGGT |
| HP1040 | TAAGTTCTAGATGACGATGGTAAAAGGGCAGAC |
| HP1714 | ATCGGAATTCTGATTGGCGATCTTCTTTTGG |
| HP1715 | TATCGGATCCATAATTGGGGTCATACTTTCAC |
| HP1801 | TATCGGATCCGTTGTTTCGGCAACGTGGTC |
| HP1832 | ATCCGCATGCACCAAGTTCATAGCAAG |
| HP1833 | ATACTGTCGACTAAACAAAAAACCCAGCTCATTAAGC |
| TGGGTTTGCGCATCACTTTATTTTGCATACTCTACGGCT | |
| CGAGTC | |
| HP2078 | ACTGAGAATTCCGTAAACACTACATTCAAAATAATCC |
| HP2079 | CATAGCAAGAGGAGGTGAAAGTATGAATACATACGAA |
| CAAATTAATAAAG | |
| HP2080 | CTTTATTAATTTGTTCGTATGTATTCATACTTTCACCTCCT |
| CTTGCTATG | |
| HP2083 | TAAGTTCTAGAGGATAAGTGAGTGTTCATTAGAAC |
| HP2104 | AATTCCTTGCCGTCAAAAAAATAAAGTGATGCTTAGCGC |
| ATCACTTTATAATTTTTTTAATCTGTTATTTAAATAGTTTA | |
| TAG | |
| HP2104rev | CTATAAACTATTTAAATAACAGATTAAAAAAATTATAAA |
| GTGATGCGCTAAGCATCACTTTATTTTTTTGACGGCAAGG | |
| AATT | |
| HP2113 | CAGCGTTCGTCCTGAGC |
| HP2164 | ATCGGAATTCTGATTGGCGATCTTCTTTTGGATGAATTGC |
| HP2165 | TATCGGATCCAGCAAAATGGAGATGAGAACCATCATAA |
| TTGG | |
| HP2227 | GGGTCTTCTTGCCGCCATCGGGAAAGC |
| HP2228 | GCTTTCCCGATGGCGGCAAGAAGACCC |
| HP2166 | TATCGGATCCGCTTCGGCAATGGTTTTAC |
| HP2238 | GTAACATTTGTTTTTTTAACATGATTG |
| HP2239 | TTCAATTAATACGACTCACTATAGGTTGTTGCTTAAA |
| AAGTGTC | |
| HP2248 | CTCCAGTCTTCACATCGGTTTG |
| HP2249 | GGTTTTGGCCGACGCTGGATCTC |
| HP2268 | TATCGGATCCATAATTGGGGTCATACTTTCACCTCCTCTT GCTATGTTACAACTTCATTGTAAATGTGTCGG |
| HP2269 | GGGTCATACTTTCACCTCCTCTTGCTATGTTACAACTTCA |
| TTGTAAATGTGTCGGAAATACTTG | |
| HP2270 | CAGTTGCGCAGCCTGAATGG |
| HP2271 | TAATACGACTCACTATAGGGAACAAACGGCGGATTGAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Korobeinikova, A.; Laalami, S.; Berthy, C.; Putzer, H. RNase Y Autoregulates Its Synthesis in Bacillus subtilis. Microorganisms 2023, 11, 1374. https://doi.org/10.3390/microorganisms11061374
AMA Style
Korobeinikova A, Laalami S, Berthy C, Putzer H. RNase Y Autoregulates Its Synthesis in Bacillus subtilis. Microorganisms. 2023; 11(6):1374. https://doi.org/10.3390/microorganisms11061374
Chicago/Turabian StyleKorobeinikova, Anna, Soumaya Laalami, Clément Berthy, and Harald Putzer. 2023. "RNase Y Autoregulates Its Synthesis in Bacillus subtilis" Microorganisms 11, no. 6: 1374. https://doi.org/10.3390/microorganisms11061374
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.