Abstract
Chlamydia infection represents an important cause for concern for public health worldwide. Chlamydial infection of the genital tract in females is mostly asymptomatic at the early stage, often manifesting as mucopurulent cervicitis, urethritis, and salpingitis at the later stage; it has been associated with female infertility, spontaneous abortion, ectopic pregnancy, and cervical cancer. As an obligate intracellular bacterium, Chlamydia depends heavily on host cells for nutrient acquisition, energy production, and cell propagation. The current review discusses various strategies utilized by Chlamydia in manipulating the cell metabolism to benefit bacterial propagation and survival through close interaction with the host cell mitochondrial and apoptotic pathway molecules.
1. Introduction
Chlamydia is an ancient infectious disease with the earliest depiction of its infection stretching as far back as 1500 BCE in the Egyptian medical compendium Ebers papyrus [1]. The genus and species designations of the organism are derivatives of the words chlamys and trachys in the Greek language, which respectively translate into “cloak” and “rough”. Ludwig Halberstädter and Stanislaus von Prowazek coined the genus name in 1907, which stemmed from their observations that specimens of conjunctiva collected from infected orangutan contained inclusions that appeared clustered around the nucleus [2]. The species designation was coined to reflect the clinical appearance of trachoma, which is characterized by roughened eyelids in patients [3,4,5]. Ensuing discoveries of the same intracytoplasmic inclusions in 1911 from children suffering from nongonococcal conjunctivitis, as well as scrapings obtained from their parents with urethritis and cervicitis, would later establish C. trachomatis as the etiologic agent of trachoma and genital infections [6]. The bacteria were at one time considered to be a virus owing to their ability to pass through filtration system and their inability to propagate on artificial growth media [7]. The debate about the pathogen’s identity was eventually ended following assignment of the infectious entity to the genus Chlamydia [7,8].
C. trachomatis is an obligate intracellular Gram-negative pathogen in the family Chlamydiaceae that predominantly infects the mucosal epithelia of the reproductive tract, leading to sexually transmitted disease in humans [9]. Alongside trichomoniasis, gonorrhea, and syphilis, genital C. trachomatis infection is one of the four most common curable sexually transmitted infections (STIs) in the world, with a worldwide incidence of more than 131 million infections every year [10]. Genital chlamydial infection is prevalent in the urban population, especially among young female adults and prostitutes [11,12]. The prevalence is also relatively higher among females with gynecological issues including infertility, endometriosis, and abnormal menstruation [13]. An increasing trend of C. trachomatis STIs has been reported worldwide in recent years, especially among young adolescents. This has raised severe public health concern as an efficient prophylactic vaccine is still not available at present. Chlamydial infections of the genital tract in females are characterized by a vast spectrum of genital tract pathologies that include mucopurulent cervicitis, urethritis, and salpingitis [9,14,15]. The silent nature of the predominant cases of infection has hampered early detection [16]. Lingering infection and its subsequent spread to the upper genital tract are linked to the development of pelvic inflammatory disease (PID), which is a known risk factor for infertility, ectopic pregnancy, and cervical cancer [17,18].
There are currently a total of 19 different C. trachomatis serovars known, distinguished by serotype characterization of the chlamydial major outer membrane protein (OmpA), and they differ considerably in terms of tissue tropism [19,20]. Serovars A, B, Ba, and C are the infectious cause of trachoma which can lead to blindness. Although trachoma is now a thing of the past in the majority of developed countries, it remains a menace in impoverished regions around the globe where there is limited access to healthcare, particularly the Middle East, Asia, and Africa. On the other hand, C. trachomatis serovars D, Da, E, F, G, Ga, H, I, Ia, J, and K infect the urogenital tract, whereas L serovars (L1, L2, L2a, and L3) invade the lymphatics and lymph nodes, resulting in lymphogranuloma venereum (LGV). C. trachomatis infection has also been linked to reactive arthritis at remote joints that often commences a few weeks following the primary site bacterial infection [21]. Certain members of the Chlamydia genus such as C. pneumoniae cause respiratory tract infections in humans, whereas other species including C. felis, C. avium, and C. abortus, despite infecting nonhuman animals as their primary host, can cause zoonotic disease in humans [22].
2. Chlamydia Metabolism
2.1. Chlamydia as an Energy Parasite
The host-restricted lifestyle of C. trachomatis is reflected in its limited genome of approximately 1.04 Mbp that is deficient in many essential genes necessary for the synthesis of metabolic cofactors, amino acids, lipids, and nucleotides. In virtually all strains of C. trachomatis characterized, a ~7.5 kbp plasmid that has been implicated in bacterial virulence is also present [23,24]. The pathogen is unable to carry out de novo biosynthesis of ATP, UTP, and GTP. Nevertheless, C. trachomatis has retained a functional CTP synthetase gene which permits the conversion of UTP to CTP [25,26]. Both Chlamydia EBs and RBs can survive under axenic conditions with defined nutrients. Whereas EBs generate ATP from the metabolism of glucose-6-phosphate, ATP, and amino acids, RBs scavenge the host ATP. This distinct ability to harness different energy sources ensures the survival of EBs in the extracellular environment prior to infection [27,28,29,30].
For decades, the bacteria in the family Chlamydiaceae were considered as exclusive “energy parasites” that feed on the host ATP to fulfill their demands for energy. This largely stemmed from the early biochemical observations that chlamydial organisms show no demonstrable succinoxidase and cytochrome c reductase activities, which pointed to the absence of several critical components of the mitochondrial respiratory chain, i.e., flavoproteins and cytochromes [31,32,33]. Subsequently, it was discovered that Chlamydia species possess a pair of nucleotide transporters, Npt1 and Npt2 (nucleoside phosphate transporters 1 and 2), allowing them to siphon nucleotides from their host. This, along with the finding that Chlamydiaceae lack the capability for de novo biosynthesis of nucleotides, with the exception of CTP, further bolstered the energy parasite hypothesis [26,34,35]. Genomic studies in the later years, however, shed a different light on the energy metabolism of Chlamydia and uncovered a slew of information on the oftentimes underappreciated metabolic capabilities of these organisms.
2.2. Glucose Metabolism in Chlamydia
The genome of C. trachomatis underwent substantial reduction in the course of its evolution as it geared toward an intracellular lifestyle, resulting in the loss of many essential genes. Despite this, C. trachomatis encodes and expresses a complete pentose phosphate pathway (PPP) and virtually all necessary enzymes to support glycolysis. While this may allow the bacterium to produce ATP and the fundamental reducing agents NADH and NADPH, it lacks a functional hexokinase gene and, therefore, draws on the host’s reserves of glucose in the form of glucose-6-phosphate (G6P) [24,36,37,38,39,40,41,42,43,44,45,46,47]. Consistent with this, experimental results indicate that the EBs and RBs differ in their requirements for energy metabolites, with G6P being the preferred substrate for EBs, whereas ATP is utilized by the RBs [29,48]. ATP is taken up by Chlamydia using a dedicated pair of ATP/ADP translocases [26,34,35]. As Chlamydia do not possess a phosphotransferase system (PTS) for the uptake of carbohydrates, G6P is instead imported into the bacteria through a sugar-phosphate/inorganic-phosphate antiporter [28,49,50,51].
C. trachomatis also possesses a complete gluconeogenic pathway and all components required for glycogen synthesis and glycogenolysis, but it remains unclear to which degree these pathways contribute to their energy metabolism. In fact, C. trachomatis appears to lack a functional carbon catabolite repression (CCR) control system; therefore, it is unable to grow well in environments enriched with gluconeogenic precursors such as glutamate, malate, α-ketoglutarate, and oxaloacetate, suggesting that these pathways are unlikely to serve as efficient input of energy [49,50,52,53].
2.3. Handicapped TCR Cycle in Chlamydia
A further limitation imposed on chlamydial energy metabolism is its fragmented tricarboxylic acid (TCA) cycle, which is devoid of citrate synthase, aconitase, and isocitrate dehydrogenase, which catalyze the first three reactions of the process. Although there is evidence that C. trachomatis can circumvent this shortfall by acquiring part of the TCA intermediates from the host, such as malate (and metabolize it to fumarate and succinate), the proteins succinate dehydrogenase subunit C and fumarate hydratase carry frameshift mutations in their coding genes (SdhC and FumC), indicating that these TCA enzymes may not operate optimally [54,55,56,57]. This evidence strongly points to a lifestyle of C. trachomatis that hinges to a significant extent on its capacity to siphon the energy metabolites from the host.
3. Manipulation of Host Mitochondria by Chlamydia
3.1. Alteration of Mitochondria Dynamics
Recently, several lines of evidence have begun to emerge suggesting that C. trachomatis is able to tip the scale of mitochondrial dynamics in favor of fusion by elevating the level of cyclic AMP (cAMP), resulting in AMP-dependent protein kinase (PKA)-dependent phosphorylation of DRP1 and its consequent inhibition. Accordingly, pharmacological inhibition of adenylate cyclase reduces intracellular cAMP, which impairs mitochondrial elongation and attenuates chlamydial growth [58,59]. Analyses of the infection-induced alteration of miRNA expression have incriminated miRNA in the regulation of p53 during chlamydial infection. miR-30c was upregulated in the infected cells, even in the presence of intense oxidative stress, which would otherwise activate p53 and its downstream target DRP1. Overexpression of both p53 and DRP1 led to severe mitochondrial fragmentation, which restricted chlamydial growth [58]. Another study suggested that Chlamydia increases mitochondrial fusion by inhibiting the activation of DRP1 and inhibits mitochondrial fission by blocking DRP1 oligomerization [60]. Silencing mitochondria fusion mediator protein or treatment with adenylate cyclase inhibitor (which diminishes mitochondrial elongation) attenuates chlamydia proliferation in cells, suggesting that modification of mitochondrial dynamics maintains a favorable environment for reproduction and growth of Chlamydia [59].
3.2. Chlamydia Upregulates HKII to Promote Mitochondria Integrity
Further evidence linking metabolic reprogramming to apoptosis regulation during chlamydial infection derives from an investigation into the interplay between hexokinase and mitochondria. In this study, it was shown that chlamydial infection triggers the phosphorylation of one of the downstream nodes of PI3K/AKT signaling cascade, 3-phosphoinositide-dependent kinase 1 (PDPK1), leading to stabilization of MYC and subsequent upregulation of hexokinase II (HKII) [61]. HKII, through its association with the mitochondrial porin voltage-dependent anion channel (VDAC), cooperates with other proteins including glucose transporter (GLUT) as well as adenine nucleotide translocator (ANT) to take part in carbohydrate metabolism by promoting the production of G6P that serves as input substrate for glycolysis, PPP, and TCA cycle [62]. Additionally, HKII-VDAC complex is involved in maintaining the outer mitochondrial membrane (OMM) integrity by interfering with the mitochondrial cell death pathway, in part due to its ability to interact with the antiapoptotic members of the BCL-2 family proteins to inhibit the mitochondrial binding of the pore-forming proapoptotic effectors BAK and BAX [63,64,65]. Consistent with the role of HKII as an important player in the regulation of apoptosis, interrupting the interaction between HKII and the mitochondria sensitizes infected cells to apoptosis [61].
3.3. Chlamydia Mediated Interference of Mitochondrial Import Machinery and Protein Composition
For some species of Chlamydia such as C. psittaci, maintaining a close contact of its inclusion with the mitochondria appears integral to its infectious cycle. Notably, disrupting the interaction of mitochondria with the C. psittaci inclusions by inhibiting kinesin, a motor protein that takes part in the movement of mitochondria in certain cell types, restricts the mobilization of mitochondria to close apposition with the inclusions, thus slowing the RB–EB conversion [66,67]. Derre et al. performed a genome-wide RNAi screen and identified two TOM subunits, i.e., Tom40 and Tom20, that are essential for C. caviae infection in Drosophila cells [68]. Knockdown of Tom40 and Tom20 decreased the infection of C. caviae in HeLa-229 cells. Interestingly, infection of Tom40-depleted cells reduced the recruitment of mitochondria to the vicinity of inclusions, which could be a plausible reason for the observed inhibition of C. caviae infection in Tom40 knockdown cells [68]. In an overexpression study of C. trachomatis genes in a yeast expression system, CT084, a conserved HKD phospholipase D (PLD)-like protein proposed to play a role in chlamydial pathogenesis, drastically impacted the growth of yeast. CT084 has been experimentally demonstrated to have mitochondrial localization in yeast, suggesting that its cytotoxicity is related to its ability to interfere with mitochondrial function [69].
Recent proteomic data have highlighted interactions between five chlamydial inclusion membrane (Inc) proteins (CT005, CT058, CT195, CT556, and CT819) components of the TIM–TOM complex [70]. The interplay between Chlamydia and the mitochondrial protein import pathway can fulfill certain functions that likely benefit its survival. For example, the intricate association of C. psittaci inclusions between its inclusions and mitochondria may be essential to rewire the mitochondrial-based pathways, possibly to facilitate the acquisition of nutrients and evasion of host cell death. Chlamydia has been known to deploy effector proteins into its host cells via the type III secretion system (T3SS) [71]. Conceivably, some of these effectors may be delivered to the mitochondria through the mitochondrial protein import system or become integrated into the mitochondria and influence the function of mitochondria. Such might be the case of C. caviae where Tom40 or Tom22 depletion attenuated bacterial infectivity but not production of ATP and apoptosis rate in infected cells, suggesting that alteration of mitochondrial protein import machinery may have an impact on other mitochondria-targeting mechanisms of survival relevance for C. caviae [68]. Employing bioinformatics screening and ectopic expression to identify chlamydial proteins with mitochondrial targeting sequence (MTS), Dimond et al. identified five chlamydial proteins (CT132, CT529, CT618, CT642, and CT647) that were localized to the mitochondria. Although the significance of these proteins in the context of bacteria–host interaction was not experimentally explored, three of these five proteins, namely, CT529, CT618, and CT642, which are putative chlamydial Inc proteins, were found to be T3SS-secreted proteins [72]. Analysis of the mitochondrial proteome from cells infected with C. trachomatis using liquid chromatography (LC) tandem mass spectrometry (MS) (LC–MS/MS) revealed mitochondrial localization of CT529 and CT618 in infected cells, as well as uncovered infection-induced changes in the mitoproteome composition, including those responsible for mitochondrial dynamics, apoptosis, and metabolism [72]. Additionally, the authors found that the chlamydial protease-like activity factor (CPAF), a secreted serine protease uniquely conserved in chlamydial organisms with a broad substrate specificity [73], and the Chlamydia protein associated with death domain (CADD), which is known to induce cell death [74], were significantly enriched in the mitochondrial proteome, suggesting that these proteins could play a role in modulating the mitochondrial processes.
4. Chlamydia and Disruption of Host Cell Metabolism
4.1. C. trachomatis Steers the Host toward Hypermetabolic State
To search for the key metabolic processes and host proteins that are essential for chlamydial growth and evaluating the suitability of these factors as drug targets in the treatment of C. trachomatis, Rother et al. [75] probed the response of HeLa cells toward C. trachomatis infection using a combined approach of gas chromatography/mass spectrometry (GC–MS), as well as small interfering RNA (siRNA). Replication of C. trachomatis was found to require several glycolytic enzymes including glucose-6-phosphate isomerase (GPI) and 6-phosphofructokinase (PFKM). Furthermore, a concomitant increase in G6P was observed, demonstrating that enhanced host cell glycolysis is essential for Chlamydia propagation. C. trachomatis also induced significant upregulation of glutamate, pyruvate, and lactate.
Additionally, RNAi screening highlighted the importance of pyruvate dehydrogenase kinase 2 (PDK2) during infection. PDK2 is a mitochondrial enzyme that regulates the activity of the pyruvate dehydrogenase kinase complex (PDC), which is responsible for oxidative decarboxylation of pyruvate to acetyl-CoA in order to enter the TCA cycle during anaerobic metabolism. Phosphorylation by PDK2 inactivates PDC; hence, more intermediates of glycolysis can be redirected to other metabolic pathways such as PPP for de novo nucleotide synthesis, as well as aerobic glycolysis, whereby pyruvate is metabolized to lactate [76]. Increased aerobic glycolysis is most commonly associated with actively proliferating cancer cells [77]. Lactate, the end-product of aerobic glycolysis, can serve as an alternative source of energy for cancer cells. Human cervix squamous carcinoma cells, for instance, are capable of utilizing lactate through oxidation to pyruvate, which can then be channeled back to glycolysis to produce ATP [78]. Thus, in a manner analogous to cancer cells, C. trachomatis steers the host to a hypermetabolic state in order to cope with increased metabolic requirements of its proliferation.
4.2. Chlamydial Propagation Requires Purine Metabolism
In keeping with the fact that C. trachomatis triggers the degradation of p53 to maintain the PPP, several PPP enzymes were identified as crucial host factors for chlamydial growth, namely, glucose-6-phosphate dehydrogenase (G6PD) and thiamine pyrophosphokinase isoform 1 (TPK1) [75]. G6PD is a key enzyme in the oxidative PPP that oxidizes G6P to produce phosphogluconolactone (PG6) [79]. TPK1, on the other hand, catalyzes the phosphorylation of thiamine into thiamine pyrophosphate (TPP), which acts as a cofactor for transketolase (TKT) of PPP [80]. TKT generates ribose-5-phosphate (R5P) in a reversible reaction belonging to the nonoxidative arm of the PPP [81]. In this way, the PPP provides the initial substrate, R5P, required for nucleotide biosynthesis. Indeed, C. trachomatis infection elevated R5P and guanosine monophosphate (GMP) in the host, indicating an increase in substrate flux toward the PPP and, subsequently, de novo purine metabolism.
Since the genome of C. trachomatis does not encode the complete enzyme paraphernalia involved in nucleotide biosynthesis including GTP, supply of nucleotides must be compensated for by the host [24]. Indeed, four additional host factors that are involved in de novo purine metabolism i.e., phosphoribosyl pyrophosphate synthetase 2 (PRPS2), phosphoribosyl pyrophosphate amidotransferase (PPAT), inosine-5-monophosphate dehydrogenase 2 (IMPDH2), and guanosine monophosphate synthetase (GMPS), were found to be significantly associated with Chlamydia propagation. Interference with these enzymes through small hairpin RNA (shRNA) in HeLa and primary ectocervical cells limited the formation of infectious C. trachomatis progeny. Notably, mice treated with the pharmacological inhibitor of IMPDH2 i.e., mycophenolate mofetil (MMF) had significantly reduced bacterial load and disease burden upon C. trachomatis infection, indicating that successful replication of C. trachomatis is tied to a functional host de novo purine metabolism [75].
4.3. Low Level of p53 Favors Chlamydial Growth
Infection with C. trachomatis is known to activate the PI3K/AKT pathway, which stimulates mouse double min 2 homolog (MDM2)-mediated degradation of the tumor suppressor protein p53 [82,83]. In addition to its well-established role in controlling the onset and progression of cancer development, p53 regulates many different effector genes and, therefore, holds sway over a variety of processes related to apoptosis, cell-cycle progression, DNA repair, cellular senescence, and metabolic adaptation [84]. In the context of metabolism, p53 negatively regulates the rate-limiting enzyme glucose-6-phosphate dehydrogenase (G6PD) of PPP, which generates NADPH required for the production of reduced glutathione (GSH), an antioxidant [85,86]. MDM2 controls the activity of p53, which stably maintains p53 at low levels under normal circumstances. MDM2 inhibits the transcriptional activation of p53. In addition, M2M2 acts as an E3 ubiquitin ligase and promotes the ubiquitination of p53, resulting in p53 nuclear export and condemning p53 to degradation by proteasome [87].
It is known that early-onset C. trachomatis infection induces the formation of ROS [88]. If left unchecked, excessive buildup of ROS is detrimental to the cells as it causes damage to DNA in the form of double-stranded breaks and telomere shortening [89,90]; both events have been observed following chlamydial infection [91,92,93]. In the event of severe DNA damage, p53 can trigger the arrest of cell-cycle progress and apoptosis [84]. This poses a threat to C. trachomatis as its survival relies on the host cell. Maintaining low levels of p53, therefore, may enable C. trachomatis to counteract the cytotoxic stress induced by DNA damage, by shifting the metabolic balance away from glycolysis to PPP to produce NADPH and nucleotides, which are crucial for antioxidant defense and promoting DNA repair. Indeed, downregulation of p53 during chlamydial infection reduced the expressions of both the cell-cycle arrest mediator p21 and the BH3-only proapoptotic protein PUMA. Furthermore, disrupting G6P activity and p53–MDM2 interaction impairs the development of C. trachomatis, attesting to the importance of p53 to metabolic reprogramming and apoptosis regulation in infected cells [82,83].
5. Chlamydia Intervenes in Host Cell Apoptosis
5.1. A General Overview of Apoptosis
There are two major pathways of apoptosis, namely, the intrinsic and extrinsic pathways, and a third mode is induced by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells (Figure 1). These signaling pathways may act in concert or independently to commit the cells to the irreversible fate of death. Irrespective of the route of initiation, the overarching endpoint for all modes of apoptosis is the activation of effector caspases 3, 6, and 7, which execute the proteolytic cleavage of numerous cellular proteins, many of these perform structural and regulatory roles, in order to prepare cells for eventual destruction [94]. The baculovirus IAP repeat (BIR) domain-containing inhibitor of apoptosis proteins (IAPs) family of proteins, notably X-chromosome-linked IAP (XIAP), cellular IAP-1 and -2, (cIAP1 and cIAP2), governs the activity of caspases by directly binding and inhibiting their activation. These proteins also possess a characteristic RING domain that allows them to interact with ubiquitin ligases; thus, they may play a role in the regulation of caspases through the ubiquitin proteasome system [95]. The second mitochondria-derived activator of caspases (SMAC/DIABLO) and serine protease high temperature requirement protein A2 (HtrA2)/OMI, which are both released from the mitochondria during apoptosis, counteract and irreversibly inactivate the IAPs via either the ubiquitin–proteasome pathway or direct proteolysis, thus clearing the way for the apoptosis cascade [96,97].
The extrinsic apoptotic pathway responds to external death stimuli through a number of transmembrane death receptors belonging to the tumor necrosis factor (TNF) superfamily, including Fas/CD95 and TNF-related apoptosis-inducing ligand (TRAIL) receptors. When engaged with their cognate ligands, for instance, TRAIL and FasL/CD95L, the death receptor trimer aggregates which recruits the Fas-associated death domain (FADD) adaptor protein via homotypical interaction of death domains (DD) [98,99]. The Fas-FADD in turn, binds the inactive initiator procaspases -8 and -10. Interactions between the death effector domain (DED) of the FADD and procaspases form the death-inducing signaling complex (DISC) wherein the procaspases dimerize and undergo proximity-induced autocatalysis to yield their active forms [100]. The mature initiator caspases process and activate the effector caspases to carry out various proteolysis events to elicit cell death. In some instances, the extrinsic pathway can convey the apoptotic cues to the intrinsic pathway. The initiator caspases orchestrate this process by cleaving the BH3-interacting domain death agonist (BID) to generate truncated BID (tBID), which then makes its way to the mitochondria to amplify apoptosis via the intrinsic pathway [101,102].
A variety of cytotoxic cellular events including organelle damage, expression of oncogenes, deprivation of cytokines and growth factors, developmental cues, and chemotherapy drugs can initiate the intrinsic pathway. When sufficiently activated, the BH3-only proteins bind and induce conformational changes in BAK and BAX to result in the formation of oligomeric pores on the OMM, leading to mitochondrial outer membrane permeabilization (MOMP). This loss of OMM integrity causes the release of apoptogenic factors from the intermembrane space into the cytosol, including cytochrome c, SMAC/DIABLO, and HtrA2/OMI [103]. Cytochrome c mediates the heptameric assembly of the apoptosome by binding to the apoptotic protease-activating factor 1 (APAF1) and dATP. Recruitment of monomeric procaspase 9 by the apoptosome and subsequent dimerization-induced activation produce the mature caspase 9 necessary to kick start the effector caspase cascade [104]. The intrinsic pathway is tightly regulated by diverse members of the BCL-2 family of proteins that are defined by the presence of one or multiple BCL-2 homology (BH) domains (BH1–BH4). These proteins are categorized into three distinct groups, i.e., prosurvival members (BCL-2, BCL-B, BCL-XL, BCL-W/BCL-2-L2, BFL1/BCL2-A1, and MCL-1), the BH3-only proapoptotic initiator proteins (BAD, BID, BIK, BIM, BMF, HK, NOXA, and PUMA), and the pore-forming proapoptotic effector proteins (BAK, BAX, and BOK) [103,105,106].
CTLs and NK cells provoke apoptosis via cytolytic molecules, i.e., perforin and granzyme B. Once released from the secretory granules of CTL or NK cell, perforin permeabilizes the target cell membrane by forming large pores to allow entry of granzyme B into the cell [107]. As with caspases, granzyme B is a serine protease that enables it to communicate with the cell death pathway in two distinct ways. It is able to directly activate the initiator caspases (8 and 10), and or the downstream effector caspases (3, 6, and 7). Additionally, it can employ an indirect approach to instigate the caspase chain reaction by cleaving the mitochondrial outer membrane permeabilization mediator BID to bring about cell killing through the intrinsic apoptosis route [108,109,110].

Figure 1.
Schematic summary of the major apoptosis pathways. Engagement of ligand (Fas/CD95L and TRAIL) with the transmembrane death receptors (Fas/CD95 and TRAIL receptors) activates the extrinsic pathway and recruits FADD. The Fas-FADD binds the procaspases 8 and 10, which leads to the formation of DISC where the procaspases mature. The active initiator caspases 8 and 10 proteolytically activate the effector caspases (3, 6, and 7) to execute apoptosis. In the intrinsic pathway, stimuli-activated BH3-only proapoptotic initiator proteins bind the pore-forming proapoptotic effector proteins BAK and BAX to induce MOMP, which results in the cytosolic release of cytochrome c, SMAC/DIABLO, and HtrA2/OMI [101]. Cytochrome c, in the presence of dATP, mediates the assembly of the monomeric APAF1 into the heptameric apoptosome complex, which processes and activates procaspase 9 into the mature caspase 9 to drive the downstream apoptosis cascade. The activity of the proapoptotic effector proteins is regulated by the pro-survival BCL-2 proteins (BCL-2, BCL-B, BCL-XL, BCL-W/BCL-2-L2, BFL1/BCL2-A1, and MCL-1), which are in turn controlled by BH3-only proapoptotic initiator proteins (BAD, BID, BIK, BIM, BMF, HK, NOXA, and PUMA). BID bridges the intrinsic and extrinsic pathway, which, upon proteolytic processing by the initiator caspases (8 and 10) into tBID, translocates to the mitochondria where it activates the intrinsic pathway. The perforin/granzyme pathway is initiated by perforin and granzyme B of CTL and NK cells. Upon delivery to the target cell, perforin oligomerizes, forming a transmembrane pore on target cells to allow cellular entry of granzyme B. Granzyme B acts as a serine protease by activating the initiator (8 and 10), and or effector (3, 6, and 7) caspases. Alternatively, granzyme B cleaves BID to tBID, and liaises with the intrinsic pathway to trigger apoptosis.
5.2. Inactivation of BAK and BAX Apoptotic Molecules
Research examining Chlamydia-infected cells has consistently shown that chlamydial infection confers resistance to various intrinsic and extrinsic proapoptotic stimuli, as well the granzyme B/perforin pathway, which exhibited characteristic inhibition of cytochrome c release [111,112,113,114,115]. The importance of mitochondria in the modulation of host cell apoptosis by Chlamydia is highlighted in experiments with Fas-expressing type I and type II cells. Rather than type I cells where apoptosis proceeds independently of mitochondria, it was found that Chlamydia species were able to protect type II cells from apoptosis, requiring a mitochondrial step to activate the effector caspases. This indicates that Chlamydia mediates the antiapoptotic actions through mechanisms that are directed toward the mitochondrial signaling pathways [116].
One likely strategy that Chlamydia uses to preserve the mitochondrial function is by blocking the activation of the proapoptotic BH3-only effector proteins BAK and BAX. In addition, Chlamydia appears to degrade BH3-only proapoptotic initiator proteins BAD, BIK, BIM, and PUMA, likely accounting for the lack of BAK and BAX activation and subsequent cytochrome c release during chlamydial infection [117,118,119,120]. Early reports identified CPAF as the chlamydial effector responsible for the cleavage of a number of these BH3-only proteins, including BIK, BIM, and PUMA. However, subsequent investigations of an extensive list of reported CPAF substrates encompassing BIK, BIM, and PUMA showed that these were likely artefacts of the experimental procedures due to post-cell-lysis degradation [121,122]. A subsequent study utilizing CPAF-deficient mutant cells lent further support to this observation, which did not produce a significant impact on cell viability [123]. However, it remains a question at this point whether CPAF represents a case of redundancy that plays a role in supporting the overall anti-apoptosis phenotype of infected cells. A recent study by Kontchou et al. provided interesting insight into the basis of chlamydial apoptosis inhibition and showed that C. trachomatis disrupts the interaction of BAK/BAX with VDAC2 to prevent their activation [124]. In this scenario, C. trachomatis blocks the release of BAK from VDAC2; as VDAC2 is known to regulate the activity of BAK by binding to BAK to restrict apoptosis [125], this inhibits the oligomerization of BAK, thus hindering the activation of apoptosis. Intriguingly, the chlamydial OmpA was found to associate with the mitochondria when overexpressed in uninfected human cells, and it protected cells from apoptosis stimulation, an effect largely mirroring C. trachomatis-infected cells, suggesting OmpA as a chlamydial antiapoptotic effector [124].
5.3. Inhibition of IAP Molecules
Chlamydia can target the IAPs to inhibit apoptosis via the upregulation of cIAP-2. This process requires the presence of other members of IAPs, namely, cIAP-1, XIAP, and survivin. Although the precise mechanism is not known, it is believed that interaction of cIAP-1, cIAP-2, XIAP, and survivin in infected cells forms a heteromeric complex that impedes the effector caspases, thereby attenuating apoptosis [126,127,128,129]. Enhanced expression of cIAP-2 seen during C. pneumoniae infection requires functional NF-κB signaling [127,128,130]. In comparison, C. trachomatis-infected cells challenged with proapoptotic stimuli did not lead to detectable NF-κB activity. Additionally, pharmacological blockade and deletion of NF-κB did not abolish the infection-induced apoptotic resistance, suggesting that NF-κB is likely dispensable for the antiapoptotic activity of C. trachomatis [129]. Nonetheless, the exact relevance of this pathway to other Chlamydia in the context of apoptosis modulation is not known.
5.4. Interference with BAD and PI3K/AKT Pathway
Chlamydia infection-associated activation of PI3K has been shown to depend on the host cell surface tyrosine kinase EphrinA2 receptor (EphA2). EphA2 attaches and mediates Chlamydia entry into host cell, wherein it remains bound to internalized bacteria and activates PI3K. ERK activation during the mid-phase of C. trachomatis infection stimulates the expression of EphA2, which is trafficked to and associated with inclusion. This inclusion-bound EphA2 activates PI3K, which in turn activates AKT. Noteworthily, EphA2 knockdown promoted cell apoptosis, indicating that EphA2-mediated signaling plays a supportive role in Chlamydia infection-induced apoptosis resistance [131]. C. trachomatis intervenes in the mitochondrial translocation of BAD in a PI3K/AKT-dependent manner. This involves activation of the PI3K/AKT pathway and subsequent phosphorylation of BAD. The phosphorylated BAD is then recruited to the chlamydial inclusion surface where it binds to a cellular protein 14-3-3β that associates with the chlamydial inclusion membrane protein IncG, thereby inhibiting the interaction between BAD and mitochondria [132]. Activated PKCδ can translocate to the mitochondria to trigger apoptosis [133,134]. C. trachomatis can sequester PKCδ similarly by diverting the enzyme from mitochondria to the vicinity of the inclusion that is enriched in diacylglycerol (DAG), possibly through a mechanism that requires the C1 domain of PKCδ [135]. In a similar fashion, Chlamydia can sequester caspase 9 into the inclusion which confers a block at the downstream caspase cascade, thus putting the brakes on apoptosis [136].
5.5. Stabilization of the Apoptosis Regulator MCL-1
Elevation of the pro-survival BCL-2 family member MCL-1 early during C. trachomatis infection has been found to be mediated via the Raf/MEK/ERK (MAPK/ERK) pathway and stabilized through the PI3K/AKT pathway [137,138]. In neutrophils infected with C. pneumoniae, activation of the Raf/MEK/ERK and PI3K/AKT pathways accompanies NF-κB-dependent release of IL-8, which further augments MCL-1 expression [138]. Accordingly, increased expression of MCL-1 blocks the release of SMAC, which would otherwise antagonize the activity of IAPs and subsequent activation of effector caspases [137]. Induction of the hypoxia-inducible factor-1α (HIF-1α) plays a role in the maintenance of MCL-1 during the initial stages of chlamydial infection, and likely involves the ERK pathway, as pharmacologically inactivating ERK signaling in infected cells with the MEK-1 inhibitor UO126 could prevent HIF-1α from degradation. At the later phase of infection, the level of HIF-1α decreases sharply, suggesting that Chlamydia employs other mechanisms to preserve the expression of MCL-1 during infection [139,140]. Indeed, a study by Fischer and colleagues showed that the chlamydial effector protein ChlaDub1 deubiquitinates MCL-1, which protects MCL-1 from proteasomal degradation. However, results obtained from cells infected with C. trachomatis ChlaDub1-null mutant did not exhibit a significant decrease in the MCL-1 protein levels. It is, therefore, possible that other redundant mechanisms to maintain the pool of MCL-1 might be at play [141]. In Hela cells persistently infected with C. psittaci, ERK1/2 pathway-dependent upregulation of MCL-1 expression similarly conferred an apoptosis resistance phenotype [142]. Kontchou et al. recently showed that infected cells deficient in MCL-1, BCL-W, and BCL-XL, as well as cells pretreated with inhibitors targeting these pro-survival proteins, remained resistant to apoptosis induction, suggesting that these proteins are not essential to chlamydial evasion of host cell apoptosis, and that the loss of MCL-1 could be compensated for by other antiapoptotic mechanisms operating in tandem during chlamydial infection [124].
5.6. Upregulation of BAG-1
Infection-mediated activation of MAPK/ERK kinase could also induce BCL-2 associated anthanogene 1 (BAG-1) expression [143]. BAG-1 is a known antiapoptotic protein capable of binding to BCL-2 to delay host cell death and activating the serine/threonine kinase Raf-1, which operates upstream of the MAPK/ERK pathway [144,145]. Inhibition of Raf-1 reduced the level of BAG-1 during chlamydial infection. Depletion of BAG-1 was correlated with the enhanced apoptosis rate of C. trachomatis-infected cells. On this basis, it has been suggested that the overall apoptosis resistance during chlamydia infection partly relies on this regulator of apoptosis [143].
6. The Influence of Chlamydia Plasmid Pgp3 on the Fate of Host Cells
6.1. Modulation of the Host Cell Autophagy and Apoptosis
Despite differing tissue tropism, most members of the family Chlamydiaceae, namely, C. trachomatis, C. muridarum, C. pneumoniae, C. psittaci, C. suis, C. caviae, C. felis, C. gallinacea, and C. pecorum, naturally bear a ~7.5 kb plasmid that is varyingly conserved (69–99%) amongst the species [146,147,148,149]. Although several isolates of C. trachomatis devoid of the plasmid have been characterized [150,151,152], the rarity of these plasmid-free variants suggests the presence of a strong selective pressure favoring the persistence of plasmid in the environment. Interestingly, Pgp3 is the only secreted chlamydial plasmid protein that has a cytosolic localization during infection, suggesting its possible role in the manipulation of host cell processes [153]. Pgp3 induces secretion of proinflammatory cytokines including interleukin-6 and interleukin-8 in epithelial cells, and plasmid presence has been associated with a severe pathological outcome in patients [13,154].
While much remains unknown concerning the functional significance of Pgp3 in the biology of Chlamydia, emerging evidence has incriminated Pgp3 as a chlamydial effector involved in the regulation of host apoptotic pathway. In a recent study, Lei et al. found that the protein high-mobility group box 1 (HMGB1) was upregulated in serum-starved HeLa cells following Pgp3 stimulation [155]. Moreover, Pgp3 triggered the expression of the BH3-containing protein Beclin-1 and the antiapoptotic BCL-2, which correlated with increased autophagic activity, specifically mitophagy, and reduced apoptotic rate in stimulated cells [155]. HMGB1 competitively displaces the binding of Beclin-1 from BCL-2 by mediating the phosphorylation of BCL-2 and binds to Beclin-1 to promote its autophagic function during nutrient starvation. Reduction of HMGB1 is known to trigger apoptosis owing to sustained Beclin-1–BCL-2 interaction [156]. Indeed, artificially inhibiting autophagy in HeLa cells stably transfected with Pgp3 decreased the expression of BCL-2 and resensitized cells to apoptosis [157].
6.2. Interaction with MDM2-p53
The role of Pgp3 became more evident when it was shown to be involved in the phosphorylation of MDM2 at serine-166 that is dependent on the PI3K pathway [158], which is essential for nuclear entry of MDM2 to interact with p53 [159]. Activation of the PI3K/AKT-mediated MDM2–p53 axis induced p53 degradation and attenuated cellular apoptosis, which could be reduced significantly by pharmacological inhibition of the MDM2–p53 interaction [158]. Chlamydia infection in HeLa epithelial cells is known to induce profound changes in the host proteome [160]. On a quest to search for the host proteins that interact with Pgp3, Li, Chen, Zhong, Wang and Zhong [153] employed isobaric tags in a high-throughput quantitative proteomics approach on Pgp3-transfected HeLa cells and uncovered a slew of proteins that were upregulated following Pgp3 expression. Among these, HMGB1 and the protein deglycase DJ-1 (otherwise known as Parkinson disease protein 7; PARK7) were significantly expressed [161]. DJ-1 exerts a plethora of roles that include cell survival and proliferation. These functions are attributed to its ability to modulate diverse cellular pathways such as PI3K/AKR and ERK1/2 [162]. DJ-1 can protect cells against the TRAIL-induced extrinsic apoptotic pathway by preventing the formation of DISC [163]. Pgp3 undermines the host apoptosis by increasing the expression of DJ-1 through the ERK1/2 signaling pathway. Accordingly, ablation of DJ-1 led to a reduction in ERK1/2 phosphorylation and predisposed Pgp3-transfected HeLa cells to apoptosis [164]. A mechanistic summary depicting the various strategies undertaken by C. trachomatis to subvert the host apoptotic machinery is put forward in Figure 2.
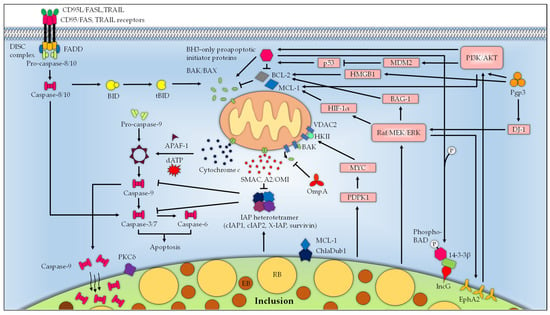
Figure 2.
The mitochondrion is the centerpiece in Chlamydia-mediated apoptosis resistance. Infection with Chlamydia activates the PI3K/AKT and Raf/MEK/ERK pathways to promote host cell survival. Co-internalization of Chlamydia EB with the host surface receptor EphA2 into the host cell triggers the activation of PI3K. Raf/MEK/ERK activation during Chlamydia infection enhances EphA2 expression, which activates AKT and sustains PI3K/AKT activation. Infection-mediated activation of PI3K/AKT leads to phosphorylation of the BH3-only protein BAD, which is diverted from the mitochondria to the inclusion protein IncG by 14-3-3β. Raf/MEK/ERK also upregulates the expression of BAG-1, which may interact with and enhance the antiapoptotic effect of BCL-2. PI3K/AKT signaling Raf/MEK/ERK stabilizes the pro-survival BCL-2 protein MCL-1. Raf/MEK/ERK-induced expression of HIF-1α maintains the MCL-1 expression. Activated PDPK1 during Chlamydia infection signals through MYC to promote the interaction between HKII and VDAC, thereby preventing BAK/BAX-induced apoptosis. Upregulation of IAPs and formation of a heterotetrameric IAP complex consisting of cIAP1, cIAP2, X-IAP, and survivin during chlamydial infection hinders the activation of effector caspases. Chlamydia infection additionally sequesters Caspase-9 within the inclusion and redirects PKCδ from the mitochondria to the inclusion to prevent apoptosis induction. Several chlamydial effector proteins contribute to the apoptosis resistance phenotype. ChlaDub1 deubiquitinates MCL-1 and protects it from proteasomal degradation, while OmpA interferes with the release of BAK from VDAC2 to inhibit BAK activation. The Chlamydia plasmid-encoded protein Pgp3, on the other hand, upregulates the Raf/MEK/ERK survival pathway through DJ-1 to promote host cell survival. In addition, Pgp3 impedes p53-mediated apoptosis and stimulates MDM2-mediated degradation of p53 by activating the PI3K/AKT pathway. Furthermore, Pgp3 upregulates the autophagic sensor HMGB1 and the antiapoptotic BCL-2 to restrict apoptosis, possibly due to autophagy-induced suppression of apoptosis.
7. Conclusions
As one of the most common bacterial STIs with increasing incidence in recent years, genital C. trachomatis infection represents a severe threat to public health afflicting the female population of childbearing age. As an obligate intracellular pathogen, C. trachomatis relies very much on its host cells to provide nutrients and other building blocks for propagation and survival. To this end, C. trachomatis deploys a well-rounded arsenal of strategies to maneuver the host metabolism and alter the cell fate. While chlamydial infections in animals and humans can be readily treated with antibiotics, identification of the tetracycline resistance gene in the swine pathogen C. suis has raised considerable concern about the potential spread of antibiotic resistance among the chlamydial species due to its zoonotic potential [165]. A more prudent way forward, therefore, would be the development of alternative and novel therapeutic regimens. Eventually, a safe and effective vaccine may be required to provide a long-term solution to these persistent pathogens. Understanding the host–pathogen interaction is, hence, essential to achieve this goal.
Author Contributions
Conceptualization, H.C.C. and C.Y.L.; writing—original draft preparation, H.C.C.; writing—review and editing, W.F.W.; supervision, L.-Y.C.; funding acquisition, S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Malaysia Ministry of Higher Education Fundamental Research Grant Scheme, grant number FRGS/1/2020/SKK0/UM/02/10 (FP050-2020).
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the Malaysia Ministry of Higher Education for funding and the University of Malaya IPPP for funding administration.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mabey, D.C.; Solomon, A.W.; Foster, A. Trachoma. Lancet 2003, 362, 223–229. [Google Scholar] [CrossRef]
- Budai, I. Chlamydia trachomatis: Milestones in clinical and microbiological diagnostics in the last hundred years: A review. Acta Microbiol. Immunol. Hung. 2007, 54, 5–22. [Google Scholar] [CrossRef]
- Kasi, P.M.; Gilani, A.I.; Ahmad, K.; Janjua, N.Z. Blinding trachoma: A disease of poverty. PLoS Med. 2004, 1, e44. [Google Scholar] [CrossRef]
- Taylor-Robinson, D. The discovery of Chlamydia trachomatis. Sex. Transm. Infect. 2017, 93, 10. [Google Scholar] [CrossRef]
- Byrne, G.I. Chlamydia uncloaked. Proc. Natl. Acad. Sci. USA 2003, 100, 8040–8042. [Google Scholar] [CrossRef]
- Darville, T. Chlamydia trachomatis infections in neonates and young children. Semin. Pediatr. Infect. Dis. 2005, 16, 235–244. [Google Scholar] [CrossRef]
- Page, L.A. Revision of the family Chlamydiaceae Rake (Rickettsiales): Unification of the psittacosis-lymphogranuloma venereum-trachoma group of organisms in the genus Chlamydia Jones, Rake and Stearns, 1945. Int. J. Syst. Evol. Microbiol. 1966, 16, 223–252. [Google Scholar] [CrossRef]
- Moulder, J.W. The relation of the psittacosis group (Chlamydiae) to bacteria and viruses. Annu. Rev. Microbiol. 1966, 20, 107–130. [Google Scholar] [CrossRef]
- Brunham, R.C.; Rey-Ladino, J. Immunology of Chlamydia infection: Implications for a Chlamydia trachomatis vaccine. Nat. Rev. Immunol. 2005, 5, 149–161. [Google Scholar] [CrossRef]
- Newman, L.; Rowley, J.; Hoorn, S.V.; Wijesooriya, N.S.; Unemo, M.; Low, N.; Stevens, G.; Gottlieb, S.; Kiarie, J.; Temmerman, M. Global Estimates of the Prevalence and Incidence of Four Curable Sexually Transmitted Infections in 2012 Based on Systematic Review and Global Reporting. PLoS ONE 2015, 10, e0143304. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ngeow, Y.F. The prevalence of sexually transmitted diseases among prostitutes in Malaysia. Sex. Transm. Infect. 1990, 66, 334–336. [Google Scholar] [CrossRef]
- Ngeow, Y.F.; Rachagan, S.P.; Ramachandran, S. Prevalence of chlamydial antibody in Malaysians. J. Clin. Pathol. 1990, 43, 400–402. [Google Scholar] [CrossRef]
- Yeow, T.C.; Wong, W.F.; Sabet, N.S.; Sulaiman, S.; Shahhosseini, F.; Tan, G.M.; Movahed, E.; Looi, C.Y.; Shankar, E.M.; Gupta, R.; et al. Prevalence of plasmid-bearing and plasmid-free Chlamydia trachomatis infection among women who visited obstetrics and gynecology clinics in Malaysia. BMC Microbiol. 2016, 16, 45. [Google Scholar] [CrossRef]
- Moss, N.J.; Ahrens, K.; Kent, C.K.; Klausner, J.D. The decline in clinical sequelae of genital Chlamydia trachomatis infection supports current control strategies. J. Infect. Dis. 2006, 193, 1336–1338. [Google Scholar] [CrossRef]
- Mackern-Oberti, J.P.; Motrich, R.D.; Breser, M.L.; Sanchez, L.R.; Cuffini, C.; Rivero, V.E. Chlamydia trachomatis infection of the male genital tract: An update. J. Reprod. Immunol. 2013, 100, 37–53. [Google Scholar] [CrossRef]
- Walker, J.; Tabrizi, S.N.; Fairley, C.K.; Chen, M.Y.; Bradshaw, C.S.; Twin, J.; Taylor, N.; Donovan, B.; Kaldor, J.M.; McNamee, K.; et al. Chlamydia trachomatis incidence and re-infection among young women—Behavioural and microbiological characteristics. PLoS ONE 2012, 7, e37778. [Google Scholar] [CrossRef]
- Hafner, L.M.; McNeilly, C. Vaccines for Chlamydia infections of the female genital tract. Future Microbiol. 2008, 3, 67–77. [Google Scholar] [CrossRef]
- Zhu, H.; Shen, Z.; Luo, H.; Zhang, W.; Zhu, X. Chlamydia trachomatis Infection-Associated Risk of Cervical Cancer: A Meta-Analysis. Medicine 2016, 95, e3077. [Google Scholar] [CrossRef]
- Caldwell, H.D.; Kromhout, J.; Schachter, J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect. Immun. 1981, 31, 1161–1176. [Google Scholar] [CrossRef]
- Bebear, C.; de Barbeyrac, B. Genital Chlamydia trachomatis infections. Clin. Microbiol. Infect. 2009, 15, 4–10. [Google Scholar] [CrossRef]
- Cheok, Y.Y.; Lee, C.Y.Q.; Cheong, H.C.; Looi, C.Y.; Wong, W.F. Chronic Inflammatory Diseases at Secondary Sites Ensuing Urogenital or Pulmonary Chlamydia Infections. Microorganisms 2020, 8, 127. [Google Scholar] [CrossRef]
- Cheong, H.C.; Lee, C.Y.Q.; Cheok, Y.Y.; Tan, G.M.Y.; Looi, C.Y.; Wong, W.F. Chlamydiaceae: Diseases in Primary Hosts and Zoonosis. Microorganisms 2019, 7, 146. [Google Scholar] [CrossRef]
- Brinkworth, A.J.; Wildung, M.R.; Carabeo, R.A. Genomewide Transcriptional Responses of Iron-Starved Chlamydia trachomatis Reveal Prioritization of Metabolic Precursor Synthesis over Protein Translation. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar] [CrossRef]
- Fisher, D.J.; Fernandez, R.E.; Maurelli, A.T. Chlamydia trachomatis transports NAD via the Npt1 ATP/ADP translocase. J. Bacteriol. 2013, 195, 3381–3386. [Google Scholar] [CrossRef]
- Wylie, J.L.; Berry, J.D.; McClarty, G. Chlamydia trachomatis CTP synthetase: Molecular characterization and developmental regulation of expression. Mol. Microbiol. 1996, 22, 631–642. [Google Scholar] [CrossRef]
- Belland, R.J.; Zhong, G.; Crane, D.D.; Hogan, D.; Sturdevant, D.; Sharma, J.; Beatty, W.L.; Caldwell, H.D. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc. Natl. Acad. Sci. USA 2003, 100, 8478–8483. [Google Scholar] [CrossRef]
- Omsland, A.; Sixt, B.S.; Horn, M.; Hackstadt, T. Chlamydial metabolism revisited: Interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol. Rev. 2014, 38, 779–801. [Google Scholar] [CrossRef]
- Omsland, A.; Sager, J.; Nair, V.; Sturdevant, D.E.; Hackstadt, T. Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc. Natl. Acad. Sci. USA 2012, 109, 19781–19785. [Google Scholar] [CrossRef]
- Grieshaber, S.; Grieshaber, N.; Yang, H.; Baxter, B.; Hackstadt, T.; Omsland, A. Impact of Active Metabolism on Chlamydia trachomatis Elementary Body Transcript Profile and Infectivity. J. Bacteriol. 2018, 200, e00065-18. [Google Scholar] [CrossRef]
- Allen, E.G.; Bovarnick, M.R. Association of reduced diphosphopyridine nucleotide cytochrome c reductase activity with meningopneumonitis virus. J. Exp. Med. 1957, 105, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.G.; Bovarnick, M.R. Enzymatic activity associated with meningopneumonitis. Ann. N. Y. Acad. Sci. 1962, 98, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.W. Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 1991, 55, 143–190. [Google Scholar] [CrossRef] [PubMed]
- Tjaden, J.; Winkler, H.H.; Schwoppe, C.; Van Der Laan, M.; Mohlmann, T.; Neuhaus, H.E. Two nucleotide transport proteins in Chlamydia trachomatis, one for net nucleoside triphosphate uptake and the other for transport of energy. J. Bacteriol. 1999, 181, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Hatch, T.P.; Al-Hossainy, E.; Silverman, J.A. Adenine nucleotide and lysine transport in Chlamydia psittaci. J. Bacteriol. 1982, 150, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.; Schofl, G.; Saluz, H.P. The Chlamydia psittaci genome: A comparative analysis of intracellular pathogens. PLoS ONE 2012, 7, e35097. [Google Scholar] [CrossRef]
- Seth-Smith, H.M.; Wanninger, S.; Bachmann, N.; Marti, H.; Qi, W.; Donati, M.; di Francesco, A.; Polkinghorne, A.; Borel, N. The Chlamydia suis Genome Exhibits High Levels of Diversity, Plasticity, and Mobile Antibiotic Resistance: Comparative Genomics of a Recent Livestock Cohort Shows Influence of Treatment Regimes. Genome Biol. Evol. 2017, 9, 750–760. [Google Scholar] [CrossRef]
- Read, T.D.; Myers, G.S.; Brunham, R.C.; Nelson, W.C.; Paulsen, I.T.; Heidelberg, J.; Holtzapple, E.; Khouri, H.; Federova, N.B.; Carty, H.A.; et al. Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): Examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 2003, 31, 2134–2147. [Google Scholar] [CrossRef]
- Azuma, Y.; Hirakawa, H.; Yamashita, A.; Cai, Y.; Rahman, M.A.; Suzuki, H.; Mitaku, S.; Toh, H.; Goto, S.; Murakami, T.; et al. Genome sequence of the cat pathogen, Chlamydophila felis. DNA Res. 2006, 13, 15–23. [Google Scholar] [CrossRef]
- Van Lent, S.; Piet, J.R.; Beeckman, D.; van der Ende, A.; Van Nieuwerburgh, F.; Bavoil, P.; Myers, G.; Vanrompay, D.; Pannekoek, Y. Full genome sequences of all nine Chlamydia psittaci genotype reference strains. J. Bacteriol. 2012, 194, 6930–6931. [Google Scholar] [CrossRef]
- Thomson, N.R.; Yeats, C.; Bell, K.; Holden, M.T.; Bentley, S.D.; Livingstone, M.; Cerdeno-Tarraga, A.M.; Harris, B.; Doggett, J.; Ormond, D.; et al. The Chlamydophila abortus genome sequence reveals an array of variable proteins that contribute to interspecies variation. Genome Res. 2005, 15, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Mojica, S.; Creasy, H.H.; Daugherty, S.; Read, T.D.; Kim, T.; Kaltenboeck, B.; Bavoil, P.; Myers, G.S. Genome sequence of the obligate intracellular animal pathogen Chlamydia pecorum E58. J. Bacteriol. 2011, 193, 3690. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Toh, H.; Hirakawa, H.; Sugii, M.; Murata, M.; Nakai, K.; Tashiro, K.; Kuhara, S.; Azuma, Y.; Shirai, M. Genome-wide analysis of Chlamydophila pneumoniae gene expression at the late stage of infection. DNA Res. 2008, 15, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Vorimore, F.; Hsia, R.C.; Huot-Creasy, H.; Bastian, S.; Deruyter, L.; Passet, A.; Sachse, K.; Bavoil, P.; Myers, G.; Laroucau, K. Isolation of a New Chlamydia species from the Feral Sacred Ibis (Threskiornis aethiopicus): Chlamydia ibidis. PLoS ONE 2013, 8, e74823. [Google Scholar] [CrossRef]
- Holzer, M.; Laroucau, K.; Creasy, H.H.; Ott, S.; Vorimore, F.; Bavoil, P.M.; Marz, M.; Sachse, K. Whole-Genome Sequence of Chlamydia gallinacea Type Strain 08-1274/3. Genome Announc. 2016, 4, e00708-16. [Google Scholar] [CrossRef]
- Taylor-Brown, A.; Spang, L.; Borel, N.; Polkinghorne, A. Culture-independent metagenomics supports discovery of uncultivable bacteria within the genus Chlamydia. Sci. Rep. 2017, 7, 10661. [Google Scholar] [CrossRef]
- Read, T.D.; Brunham, R.C.; Shen, C.; Gill, S.R.; Heidelberg, J.F.; White, O.; Hickey, E.K.; Peterson, J.; Utterback, T.; Berry, K.; et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000, 28, 1397–1406. [Google Scholar] [CrossRef]
- Skipp, P.J.; Hughes, C.; McKenna, T.; Edwards, R.; Langridge, J.; Thomson, N.R.; Clarke, I.N. Quantitative Proteomics of the Infectious and Replicative Forms of Chlamydia trachomatis. PLoS ONE 2016, 11, e0149011. [Google Scholar] [CrossRef]
- Iliffe-Lee, E.R.; McClarty, G. Glucose metabolism in Chlamydia trachomatis: The ‘energy parasite’ hypothesis revisited. Mol. Microbiol. 1999, 33, 177–187. [Google Scholar] [CrossRef]
- Iliffe-Lee, E.R.; McClarty, G. Regulation of carbon metabolism in Chlamydia trachomatis. Mol. Microbiol. 2000, 38, 20–30. [Google Scholar] [CrossRef]
- Schwoppe, C.; Winkler, H.H.; Neuhaus, H.E. Properties of the glucose-6-phosphate transporter from Chlamydia pneumoniae (HPTcp) and the glucose-6-phosphate sensor from Escherichia coli (UhpC). J. Bacteriol. 2002, 184, 2108–2115. [Google Scholar] [CrossRef] [PubMed]
- Gorke, B.; Stulke, J. Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nat. Rev. Microbiol. 2008, 6, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.L.; Chiu, K.; Stephens, R.S. Chlamydia trachomatis lacks an adaptive response to changes in carbon source availability. Infect. Immun. 2004, 72, 4286–4289. [Google Scholar] [CrossRef] [PubMed]
- Mehlitz, A.; Eylert, E.; Huber, C.; Lindner, B.; Vollmuth, N.; Karunakaran, K.; Goebel, W.; Eisenreich, W.; Rudel, T. Metabolic adaptation of Chlamydia trachomatis to mammalian host cells. Mol. Microbiol. 2017, 103, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.R.; Holden, M.T.; Carder, C.; Lennard, N.; Lockey, S.J.; Marsh, P.; Skipp, P.; O’Connor, C.D.; Goodhead, I.; Norbertzcak, H.; et al. Chlamydia trachomatis: Genome sequence analysis of lymphogranuloma venereum isolates. Genome Res. 2008, 18, 161–171. [Google Scholar] [CrossRef]
- Lancaster, C.R. The di-heme family of respiratory complex II enzymes. Biochim. Biophys. Acta 2013, 1827, 679–687. [Google Scholar] [CrossRef]
- Cecchini, G. Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Reimer, A.; Sharan, M.; Kozjak-Pavlovic, V.; Eulalio, A.; Prusty, B.K.; Fraunholz, M.; Karunakaran, K.; Rudel, T. Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J. Cell Biol. 2017, 216, 1071–1089. [Google Scholar] [CrossRef]
- Kurihara, Y.; Itoh, R.; Shimizu, A.; Walenna, N.F.; Chou, B.; Ishii, K.; Soejima, T.; Fujikane, A.; Hiromatsu, K. Chlamydia trachomatis targets mitochondrial dynamics to promote intracellular survival and proliferation. Cell. Microbiol. 2019, 21, e12962. [Google Scholar] [CrossRef]
- Yang, Y.; Lei, W.; Zhao, L.; Wen, Y.; Li, Z. Insights Into Mitochondrial Dynamics in Chlamydial Infection. Front. Cell. Infect. Microbiol. 2022, 12, 835181. [Google Scholar] [CrossRef]
- Al-Zeer, M.A.; Xavier, A.; Abu Lubad, M.; Sigulla, J.; Kessler, M.; Hurwitz, R.; Meyer, T.F. Chlamydia trachomatis Prevents Apoptosis Via Activation of PDPK1-MYC and Enhanced Mitochondrial Binding of Hexokinase II. EBioMedicine 2017, 23, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [PubMed]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 2004, 16, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B. Hexokinase II: The integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 2003, 10, 1535–1551. [Google Scholar] [CrossRef]
- Matsumoto, A.; Bessho, H.; Uehira, K.; Suda, T. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J. Electron. Microsc. 1991, 40, 356–363. [Google Scholar]
- Peterson, E.M.; de la Maza, L.M. Chlamydia parasitism: Ultrastructural characterization of the interaction between the chlamydial cell envelope and the host cell. J. Bacteriol. 1988, 170, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Derre, I.; Pypaert, M.; Dautry-Varsat, A.; Agaisse, H. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog. 2007, 3, e155. [Google Scholar] [CrossRef]
- Sisko, J.L.; Spaeth, K.; Kumar, Y.; Valdivia, R.H. Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Mol. Microbiol. 2006, 60, 51–66. [Google Scholar] [CrossRef]
- Mirrashidi, K.M.; Elwell, C.A.; Verschueren, E.; Johnson, J.R.; Frando, A.; Von Dollen, J.; Rosenberg, O.; Gulbahce, N.; Jang, G.; Johnson, T.; et al. Global Mapping of the Inc-Human Interactome Reveals that Retromer Restricts Chlamydia Infection. Cell Host Microbe 2015, 18, 109–121. [Google Scholar] [CrossRef]
- Betts-Hampikian, H.J.; Fields, K.A. The Chlamydial Type III Secretion Mechanism: Revealing Cracks in a Tough Nut. Front. Microbiol. 2010, 1, 114. [Google Scholar] [CrossRef] [PubMed]
- Dimond, Z.; Bauler, L.D.; Zhang, Y.; Carmody, A.; Hackstadt, T. Chlamydia trachomatis Alters Mitochondrial Protein Composition and Secretes Effector Proteins That Target Mitochondria. mSphere 2022, 7, e0042322. [Google Scholar] [CrossRef] [PubMed]
- Bednar, M.M.; Jorgensen, I.; Valdivia, R.H.; McCafferty, D.G. Chlamydia protease-like activity factor (CPAF): Characterization of proteolysis activity in vitro and development of a nanomolar affinity CPAF zymogen-derived inhibitor. Biochemistry 2011, 50, 7441–7443. [Google Scholar] [CrossRef]
- Stenner-Liewen, F.; Liewen, H.; Zapata, J.M.; Pawlowski, K.; Godzik, A.; Reed, J.C. CADD, a Chlamydia protein that interacts with death receptors. J. Biol. Chem. 2002, 277, 9633–9636. [Google Scholar] [CrossRef]
- Rother, M.; Gonzalez, E.; da Costa, A.R.T.; Wask, L.; Gravenstein, I.; Pardo, M.; Pietzke, M.; Gurumurthy, R.K.; Angermann, J.; Laudeley, R.; et al. Combined Human Genome-wide RNAi and Metabolite Analyses Identify IMPDH as a Host-Directed Target against Chlamydia Infection. Cell Host Microbe 2018, 23, 661–671.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Tian, W.N.; Braunstein, L.D.; Pang, J.; Stuhlmeier, K.M.; Xi, Q.C.; Tian, X.; Stanton, R.C. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 1998, 273, 10609–10617. [Google Scholar] [CrossRef]
- Nosaka, K.; Onozuka, M.; Kakazu, N.; Hibi, S.; Nishimura, H.; Nishino, H.; Abe, T. Isolation and characterization of a human thiamine pyrophosphokinase cDNA. Biochim. Biophys. Acta 2001, 1517, 293–297. [Google Scholar] [CrossRef]
- Boros, L.G.; Puigjaner, J.; Cascante, M.; Lee, W.N.; Brandes, J.L.; Bassilian, S.; Yusuf, F.I.; Williams, R.D.; Muscarella, P.; Melvin, W.S.; et al. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997, 57, 4242–4248. [Google Scholar] [PubMed]
- Siegl, C.; Prusty, B.K.; Karunakaran, K.; Wischhusen, J.; Rudel, T. Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Rep. 2014, 9, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Rother, M.; Kerr, M.C.; Al-Zeer, M.A.; Abu-Lubad, M.; Kessler, M.; Brinkmann, V.; Loewer, A.; Meyer, T.F. Chlamydia infection depends on a functional MDM2-p53 axis. Nat. Commun. 2014, 5, 5201. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Schwarzl, S.M.; Smith, J.; Osieka, R. Role of glucose-6-phosphate dehydrogenase for oxidative stress and apoptosis. Cell Death Differ. 2006, 13, 527–528. [Google Scholar] [CrossRef]
- Chene, P. Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [Google Scholar] [CrossRef]
- Boncompain, G.; Schneider, B.; Delevoye, C.; Kellermann, O.; Dautry-Varsat, A.; Subtil, A. Production of reactive oxygen species is turned on and rapidly shut down in epithelial cells infected with Chlamydia trachomatis. Infect. Immun. 2010, 78, 80–87. [Google Scholar] [CrossRef]
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE 2014, 9, e110963. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Chumduri, C.; Gurumurthy, R.K.; Zadora, P.K.; Mi, Y.; Meyer, T.F. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell Host Microbe 2013, 13, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Gurumurthy, R.K.; Zadora, P.K.; Meyer, T.F.; Chumduri, C. Chlamydia trachomatis Inhibits Homologous Recombination Repair of DNA Breaks by Interfering with PP2A Signaling. mBio 2018, 9, e01465-18. [Google Scholar] [CrossRef] [PubMed]
- Padberg, I.; Janssen, S.; Meyer, T.F. Chlamydia trachomatis inhibits telomeric DNA damage signaling via transient hTERT upregulation. Int. J. Med. Microbiol. 2013, 303, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef]
- Vaux, D.L.; Silke, J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 287–297. [Google Scholar] [CrossRef]
- Yang, Q.H.; Church-Hajduk, R.; Ren, J.; Newton, M.L.; Du, C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003, 17, 1487–1496. [Google Scholar] [CrossRef]
- Creagh, E.M.; Murphy, B.M.; Duriez, P.J.; Duckett, C.S.; Martin, S.J. Smac/Diablo antagonizes ubiquitin ligase activity of inhibitor of apoptosis proteins. J. Biol. Chem. 2004, 279, 26906–26914. [Google Scholar] [CrossRef]
- Caulfield, A.J.; Lathem, W.W. Disruption of fas-fas ligand signaling, apoptosis, and innate immunity by bacterial pathogens. PLoS Pathog. 2014, 10, e1004252. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- MacKenzie, S.H.; Clark, A.C. Death by caspase dimerization. Adv. Exp. Med. Biol. 2012, 747, 55–73. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Milhas, D.; Cuvillier, O.; Therville, N.; Clave, P.; Thomsen, M.; Levade, T.; Benoist, H.; Segui, B. Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis. J. Biol. Chem. 2005, 280, 19836–19842. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Timmer, J.; Sperandio, S.; Salvesen, G.S. The apoptosome activates caspase-9 by dimerization. Mol. Cell 2006, 22, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Akey, C.W. Apoptosome structure, assembly, and procaspase activation. Structure 2013, 21, 501–515. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Goping, I.S.; Barry, M.; Liston, P.; Sawchuk, T.; Constantinescu, G.; Michalak, K.M.; Shostak, I.; Roberts, D.L.; Hunter, A.M.; Korneluk, R.; et al. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity 2003, 18, 355–365. [Google Scholar] [CrossRef]
- Barry, M.; Heibein, J.A.; Pinkoski, M.J.; Lee, S.F.; Moyer, R.W.; Green, D.R.; Bleackley, R.C. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol. Cell. Biol. 2000, 20, 3781–3794. [Google Scholar] [CrossRef]
- Pinkoski, M.J.; Waterhouse, N.J.; Heibein, J.A.; Wolf, B.B.; Kuwana, T.; Goldstein, J.C.; Newmeyer, D.D.; Bleackley, R.C.; Green, D.R. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J. Biol. Chem. 2001, 276, 12060–12067. [Google Scholar] [CrossRef]
- Fan, T.; Lu, H.; Hu, H.; Shi, L.; McClarty, G.A.; Nance, D.M.; Greenberg, A.H.; Zhong, G. Inhibition of apoptosis in chlamydia-infected cells: Blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 1998, 187, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Rajalingam, K.; Al-Younes, H.; Muller, A.; Meyer, T.F.; Szczepek, A.J.; Rudel, T. Epithelial cells infected with Chlamydophila pneumoniae (Chlamydia pneumoniae) are resistant to apoptosis. Infect. Immun. 2001, 69, 7880–7888. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Xiao, Y.; Huang, Y.; McClarty, G.; Zhong, G. Chlamydia-infected cells continue to undergo mitosis and resist induction of apoptosis. Infect. Immun. 2004, 72, 451–460. [Google Scholar] [CrossRef]
- Fischer, S.F.; Schwarz, C.; Vier, J.; Hacker, G. Characterization of antiapoptotic activities of Chlamydia pneumoniae in human cells. Infect. Immun. 2001, 69, 7121–7129. [Google Scholar] [CrossRef] [PubMed]
- Airenne, S.; Surcel, H.M.; Tuukkanen, J.; Leinonen, M.; Saikku, P. Chlamydia pneumoniae inhibits apoptosis in human epithelial and monocyte cell lines. Scand. J. Immunol. 2002, 55, 390–398. [Google Scholar] [CrossRef]
- Fischer, S.F.; Harlander, T.; Vier, J.; Hacker, G. Protection against CD95-induced apoptosis by Chlamydial infection at a mitochondrial step. Infect. Immun. 2004, 72, 1107–1115. [Google Scholar] [CrossRef]
- Fischer, S.F.; Vier, J.; Kirschnek, S.; Klos, A.; Hess, S.; Ying, S.; Hacker, G. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J. Exp. Med. 2004, 200, 905–916. [Google Scholar] [CrossRef]
- Zhong, Y.; Weininger, M.; Pirbhai, M.; Dong, F.; Zhong, G. Inhibition of staurosporine-induced activation of the proapoptotic multidomain Bcl-2 proteins Bax and Bak by three invasive chlamydial species. J. Infect. 2006, 53, 408–414. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhong, Y.; Greene, W.; Dong, F.; Zhong, G. Chlamydia trachomatis infection inhibits both Bax and Bak activation induced by staurosporine. Infect. Immun. 2004, 72, 5470–5474. [Google Scholar] [CrossRef]
- Dong, F.; Pirbhai, M.; Xiao, Y.; Zhong, Y.; Wu, Y.; Zhong, G. Degradation of the proapoptotic proteins Bik, Puma, and Bim with Bcl-2 domain 3 homology in Chlamydia trachomatis-infected cells. Infect. Immun. 2005, 73, 1861–1864. [Google Scholar] [CrossRef]
- Jorgensen, I.; Bednar, M.M.; Amin, V.; Davis, B.K.; Ting, J.P.; McCafferty, D.G.; Valdivia, R.H. The Chlamydia protease CPAF regulates host and bacterial proteins to maintain pathogen vacuole integrity and promote virulence. Cell Host Microbe 2011, 10, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Pirbhai, M.; Dong, F.; Zhong, Y.; Pan, K.Z.; Zhong, G. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 2006, 281, 31495–31501. [Google Scholar] [CrossRef] [PubMed]
- Snavely, E.A.; Kokes, M.; Dunn, J.D.; Saka, H.A.; Nguyen, B.D.; Bastidas, R.J.; McCafferty, D.G.; Valdivia, R.H. Reassessing the role of the secreted protease CPAF in Chlamydia trachomatis infection through genetic approaches. Pathog. Dis. 2014, 71, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Kontchou, C.W.; Gentle, I.E.; Weber, A.; Schoeniger, A.; Edlich, F.; Hacker, G. Chlamydia trachomatis inhibits apoptosis in infected cells by targeting the pro-apoptotic proteins Bax and Bak. Cell Death Differ. 2022, 29, 2046–2059. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Rajalingam, K.; Sharma, M.; Paland, N.; Hurwitz, R.; Thieck, O.; Oswald, M.; Machuy, N.; Rudel, T. IAP-IAP complexes required for apoptosis resistance of C. trachomatis-infected cells. PLoS Pathog. 2006, 2, e114. [Google Scholar] [CrossRef] [PubMed]
- Paland, N.; Rajalingam, K.; Machuy, N.; Szczepek, A.; Wehrl, W.; Rudel, T. NF-kappaB and inhibitor of apoptosis proteins are required for apoptosis resistance of epithelial cells persistently infected with Chlamydophila pneumoniae. Cell. Microbiol. 2006, 8, 1643–1655. [Google Scholar] [CrossRef]
- Wahl, C.; Maier, S.; Marre, R.; Essig, A. Chlamydia pneumoniae induces the expression of inhibitor of apoptosis 2 (c-IAP2) in a human monocytic cell line by an NF-kappaB-dependent pathway. Int. J. Med. Microbiol. 2003, 293, 377–381. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhong, Y.; Su, H.; Zhou, Z.; Chiao, P.; Zhong, G. NF-kappa B activation is not required for Chlamydia trachomatis inhibition of host epithelial cell apoptosis. J. Immunol. 2005, 174, 1701–1708. [Google Scholar] [CrossRef]
- Wahl, C.; Oswald, F.; Simnacher, U.; Weiss, S.; Marre, R.; Essig, A. Survival of Chlamydia pneumoniae-infected Mono Mac 6 cells is dependent on NF-kappaB binding activity. Infect. Immun. 2001, 69, 7039–7045. [Google Scholar] [CrossRef]
- Subbarayal, P.; Karunakaran, K.; Winkler, A.C.; Rother, M.; Gonzalez, E.; Meyer, T.F.; Rudel, T. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 2015, 11, e1004846. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, P.; Welter-Stahl, L.; Ying, S.; Hansen, J.; Hacker, G.; Darville, T.; Ojcius, D.M. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog. 2006, 2, e45. [Google Scholar] [CrossRef] [PubMed]
- Matassa, A.A.; Carpenter, L.; Biden, T.J.; Humphries, M.J.; Reyland, M.E. PKCdelta is required for mitochondrial-dependent apoptosis in salivary epithelial cells. J. Biol. Chem. 2001, 276, 29719–29728. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Mishra, N.C.; Sun, X.; Bharti, A.; Kharbanda, S.; Saxena, S.; Kufe, D. Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ. 2001, 12, 465–470. [Google Scholar] [PubMed]
- Tse, S.M.; Mason, D.; Botelho, R.J.; Chiu, B.; Reyland, M.; Hanada, K.; Inman, R.D.; Grinstein, S. Accumulation of diacylglycerol in the Chlamydia inclusion vacuole: Possible role in the inhibition of host cell apoptosis. J. Biol. Chem. 2005, 280, 25210–25215. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Shirai, M.; Aziz, M.A.; Ushirokita, R.; Kubota, S.; Suzuki, H.; Azuma, Y. An epistatic effect of apaf-1 and caspase-9 on chlamydial infection. Apoptosis 2015, 20, 1271–1280. [Google Scholar] [CrossRef]
- Rajalingam, K.; Sharma, M.; Lohmann, C.; Oswald, M.; Thieck, O.; Froelich, C.J.; Rudel, T. Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PLoS ONE 2008, 3, e3102. [Google Scholar] [CrossRef]
- Sarkar, A.; Moller, S.; Bhattacharyya, A.; Behnen, M.; Rupp, J.; van Zandbergen, G.; Solbach, W.; Laskay, T. Mechanisms of apoptosis inhibition in Chlamydia pneumoniae-infected neutrophils. Int. J. Med. Microbiol. 2015, 305, 493–500. [Google Scholar] [CrossRef]
- Sharma, M.; Machuy, N.; Bohme, L.; Karunakaran, K.; Maurer, A.P.; Meyer, T.F.; Rudel, T. HIF-1alpha is involved in mediating apoptosis resistance to Chlamydia trachomatis-infected cells. Cell. Microbiol. 2011, 13, 1573–1585. [Google Scholar] [CrossRef]
- Rupp, J.; Gieffers, J.; Klinger, M.; van Zandbergen, G.; Wrase, R.; Maass, M.; Solbach, W.; Deiwick, J.; Hellwig-Burgel, T. Chlamydia pneumoniae directly interferes with HIF-1alpha stabilization in human host cells. Cell. Microbiol. 2007, 9, 2181–2191. [Google Scholar] [CrossRef]
- Fischer, A.; Harrison, K.S.; Ramirez, Y.; Auer, D.; Chowdhury, S.R.; Prusty, B.K.; Sauer, F.; Dimond, Z.; Kisker, C.; Hefty, P.S.; et al. Chlamydia trachomatis-containing vacuole serves as deubiquitination platform to stabilize Mcl-1 and to interfere with host defense. eLife 2017, 6, e21465. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, C.; Wen, Y.; Hu, Y.; Xie, Y.; Xu, M.; Liang, M.; Liu, W.; Liu, L.; Wu, Y. ERK1/2 and the Bcl-2 Family Proteins Mcl-1, tBid, and Bim Are Involved in Inhibition of Apoptosis during Persistent Chlamydia psittaci Infection. Inflammation 2018, 41, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Kun, D.; Xiang-Lin, C.; Ming, Z.; Qi, L. Chlamydia inhibit host cell apoptosis by inducing Bag-1 via the MAPK/ERK survival pathway. Apoptosis 2013, 18, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.G.; Takayama, S.; Rapp, U.R.; Reed, J.C. Bcl-2 interacting protein, BAG-1, binds to and activates the kinase Raf-1. Proc. Natl. Acad. Sci. USA 1996, 93, 7063–7068. [Google Scholar] [CrossRef]
- Takayama, S.; Sato, T.; Krajewski, S.; Kochel, K.; Irie, S.; Milian, J.A.; Reed, J.C. Cloning and functional analysis of BAG-1: A novel Bcl-2-binding protein with anti-cell death activity. Cell 1995, 80, 279–284. [Google Scholar] [CrossRef]
- Rockey, D.D. Unraveling the basic biology and clinical significance of the chlamydial plasmid. J. Exp. Med. 2011, 208, 2159–2162. [Google Scholar] [CrossRef]
- Jelocnik, M.; Bachmann, N.L.; Kaltenboeck, B.; Waugh, C.; Woolford, L.; Speight, K.N.; Gillett, A.; Higgins, D.P.; Flanagan, C.; Myers, G.S.; et al. Genetic diversity in the plasticity zone and the presence of the chlamydial plasmid differentiates Chlamydia pecorum strains from pigs, sheep, cattle, and koalas. BMC Genom. 2015, 16, 893. [Google Scholar] [CrossRef]
- Jelocnik, M.; Bachmann, N.L.; Seth-Smith, H.; Thomson, N.R.; Timms, P.; Polkinghorne, A.M. Molecular characterisation of the Chlamydia pecorum plasmid from porcine, ovine, bovine, and koala strains indicates plasmid-strain co-evolution. PeerJ 2016, 4, e1661. [Google Scholar] [CrossRef]
- Pawlikowska-Warych, M.; Sliwa-Dominiak, J.; Deptula, W. Chlamydial plasmids and bacteriophages. Acta Biochim. Pol. 2015, 62, 1–6. [Google Scholar] [CrossRef]
- Stothard, D.R.; Williams, J.A.; Van Der Pol, B.; Jones, R.B. Identification of a Chlamydia trachomatis serovar E urogenital isolate which lacks the cryptic plasmid. Infect. Immun. 1998, 66, 6010–6013. [Google Scholar] [CrossRef]
- Farencena, A.; Comanducci, M.; Donati, M.; Ratti, G.; Cevenini, R. Characterization of a new isolate of Chlamydia trachomatis which lacks the common plasmid and has properties of biovar trachoma. Infect. Immun. 1997, 65, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.M.; Markoff, B.A.; Schachter, J.; de la Maza, L.M. The 7.5-kb plasmid present in Chlamydia trachomatis is not essential for the growth of this microorganism. Plasmid 1990, 23, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Chen, D.; Zhong, Y.M.; Wang, S.P.; Zhong, G.M. The chlamydial plasmid-encoded protein pgp3 Is secreted into the cytosol of chlamydia-infected cells. Infect. Immun. 2008, 76, 3415–3428. [Google Scholar] [CrossRef]
- Cheong, H.C.; Cheok, Y.Y.; Chan, Y.T.; Tang, T.F.; Sulaiman, S.; Looi, C.Y.; Gupta, R.; Arulanandam, B.; Chang, L.Y.; Wong, W.F. Chlamydia trachomatis plasmid-encoding Pgp3 protein induces secretion of distinct inflammatory signatures from HeLa cervical epithelial cells. BMC Microbiol. 2023, 23, 58. [Google Scholar] [CrossRef]
- Lei, W.; Li, Q.; Su, S.; Bu, J.; Huang, Q.; Li, Z. Chlamydia trachomatis plasmid-encoded protein pORF5 protects mitochondrial function by inducing mitophagy and increasing HMGB1 expression. Pathog. Dis. 2017, 75, ftx111. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Zhao, Y.; Yang, X.; Su, S.; Wen, Y.; Chen, H.; Zhou, Z.; Huang, Q.; Li, Z. Chlamydia trachomatis pORF5 plasmid-encoded protein regulates autophagy and apoptosis of HeLa cells. Biotechnol. Biotechnol. Equip. 2019, 33, 1269–1279. [Google Scholar] [CrossRef]
- Zou, Y.; Lei, W.; Su, S.; Bu, J.; Zhu, S.; Huang, Q.; Li, Z. Chlamydia trachomatis plasmid-encoded protein Pgp3 inhibits apoptosis via the PI3K-AKT-mediated MDM2-p53 axis. Mol. Cell. Biochem. 2018, 452, 167–176. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Tan, G.M.; Lim, H.J.; Yeow, T.C.; Movahed, E.; Looi, C.Y.; Gupta, R.; Arulanandam, B.P.; Abu Bakar, S.; Sabet, N.S.; Chang, L.Y.; et al. Temporal proteomic profiling of Chlamydia trachomatis-infected HeLa-229 human cervical epithelial cells. Proteomics 2016, 16, 1347–1360. [Google Scholar] [CrossRef]
- Zou, Y.; Dai, W.; Lei, W.; Su, S.; Huang, Q.; Zhou, Z.; Chen, C.; Li, Z. Identification of proteins interacting with pORF5 in the pathogenesis of C. trachomatis. Am. J. Transl. Res. 2018, 10, 1633–1647. [Google Scholar]
- Oh, S.E.; Mouradian, M.M. Regulation of Signal Transduction by DJ-1. Adv. Exp. Med. Biol. 2017, 1037, 97–131. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Ren, H.; Wang, Y.; Fei, E.; Wang, H.; Wang, G. DJ-1 inhibits TRAIL-induced apoptosis by blocking pro-caspase-8 recruitment to FADD. Oncogene 2012, 31, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Shu, M.; Gong, S.; Wen, Y.; He, B.; Su, S.; Li, Z. Antiapoptotic activity of Chlamydia trachomatis Pgp3 protein involves activation of the ERK1/2 pathway mediated by upregulation of DJ-1 protein. Pathog. Dis. 2019, 77, ftaa003. [Google Scholar] [CrossRef] [PubMed]
- Bommana, S.; Polkinghorne, A. Mini Review: Antimicrobial Control of Chlamydial Infections in Animals: Current Practices and Issues. Front. Microbiol. 2019, 10, 113. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).