
Linking Groundwater to Surface Discharge Ecosystems: Archaeal, Bacterial, and Eukaryotic Community Diversity and Structure in Quebec (Canada)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Sampling
2.2. Geochemical Analyses
2.3. DNA Extraction and Illumina Sequencing
2.4. Sequence Analyses
2.5. Statistical Analyses
3. Results
3.1. Microbial Gene Diversity
3.2. Microbial α-Diversity Indices and Comparison
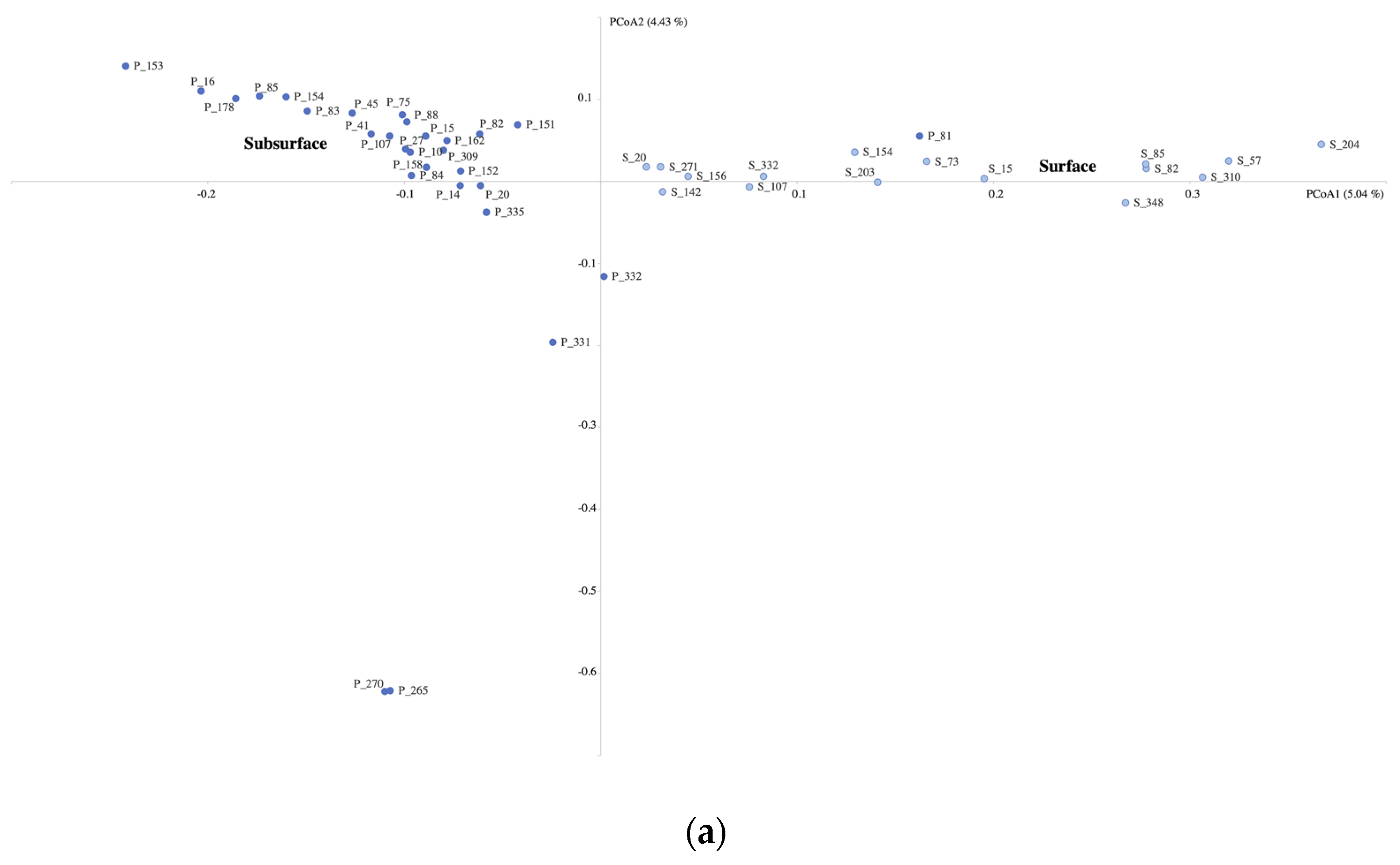
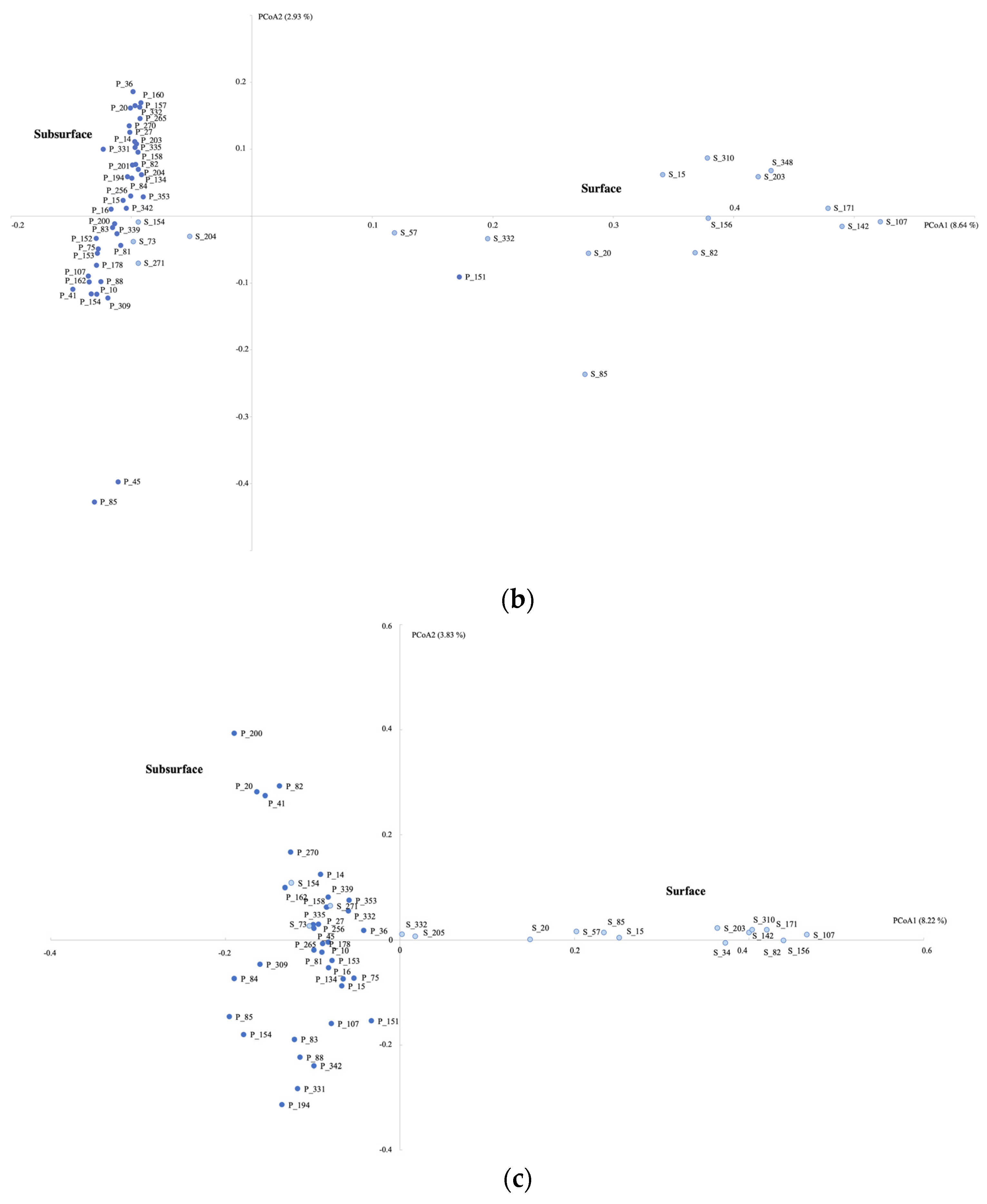
3.3. Microbial Community Composition
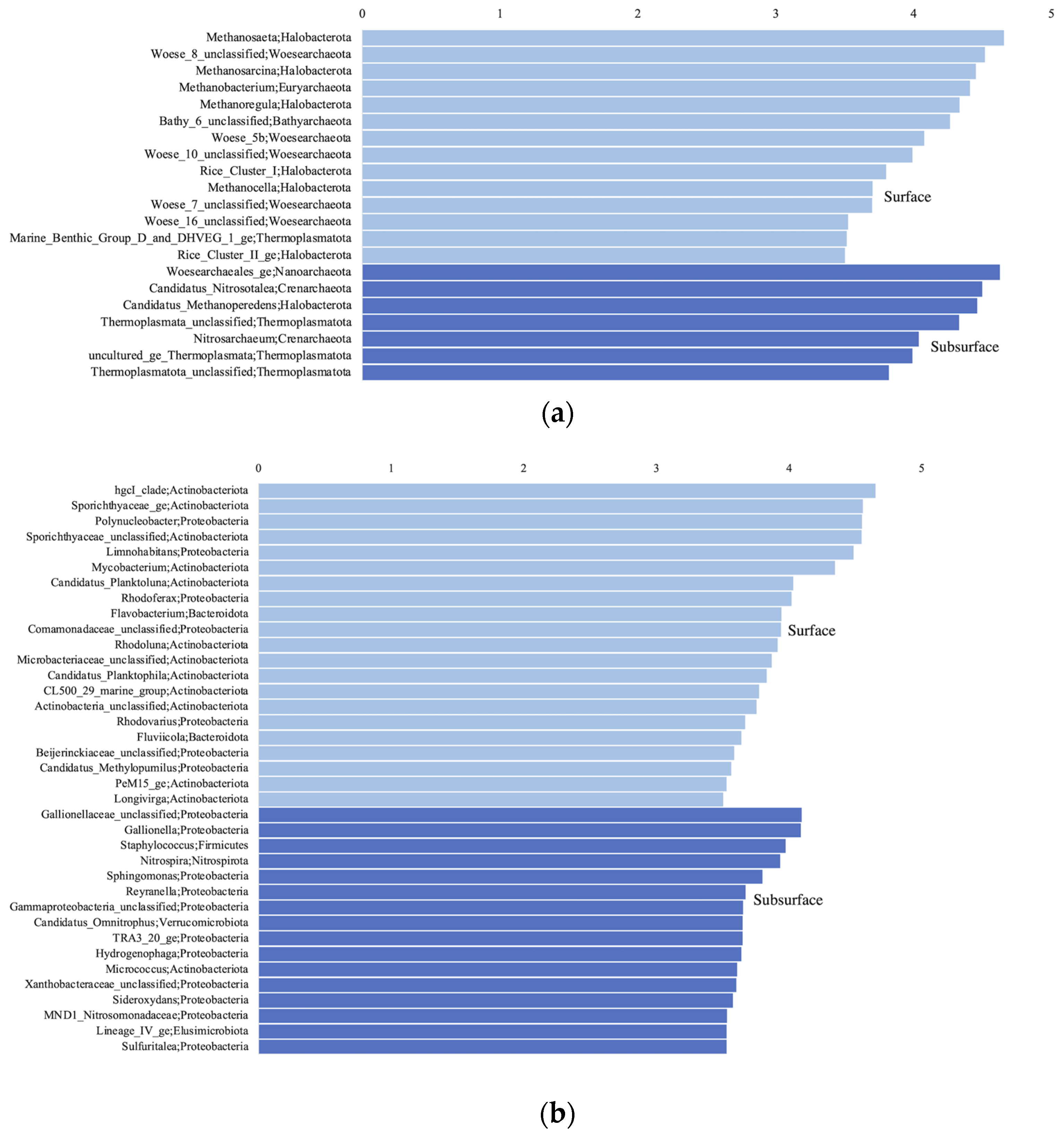
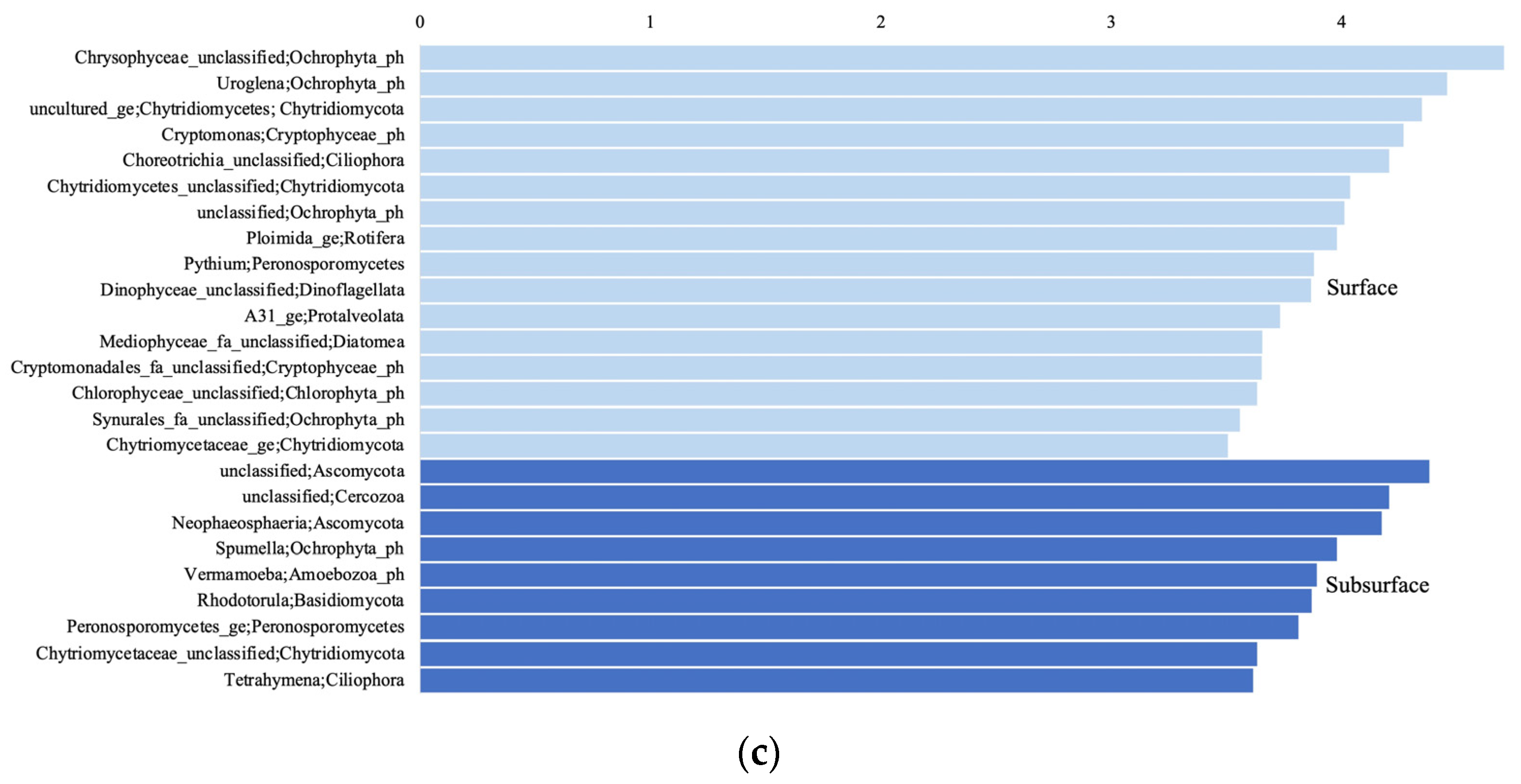
3.4. Shared and Unique Taxa in the Surface and Subsurface Samples
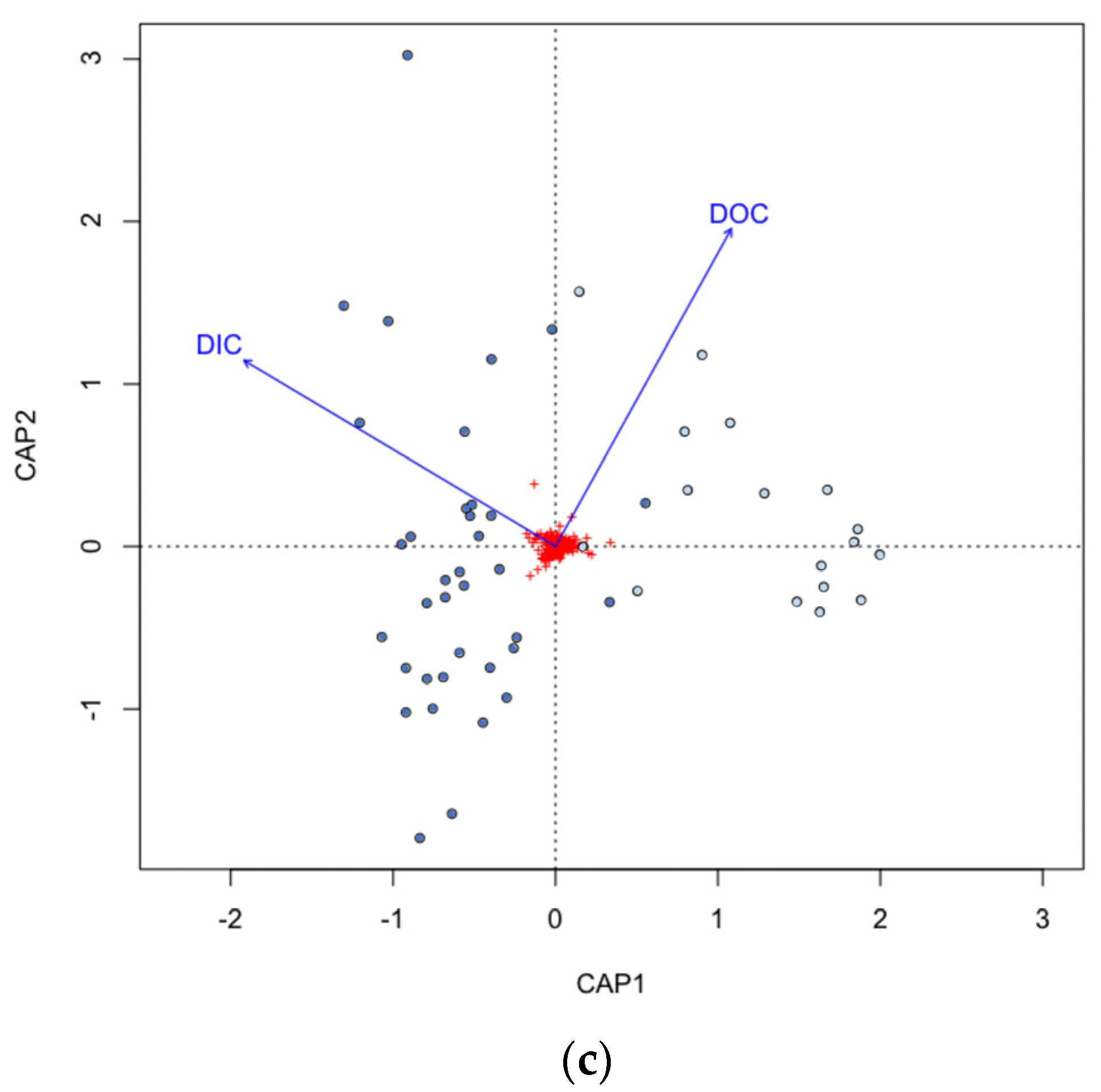
3.5. Microbial Community Correlation with Environmental Parameters
3.6. Contribution of Groundwater Microbial Communities to the Surface Communities
4. Discussion
4.1. Microbial Community Differences between Subsurface and Surface Aquatic Habitats
4.2. Distinguishing Microbial Taxa in Subsurface and Surface Aquatic Environments
4.3. Links between Subsurface and Surface Aquatic Microbial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fillinger, L.; Hug, K.; Griebler, C. Aquifer recharge viewed through the lens of microbial community ecology: Initial disturbance response, and impacts of species sorting versus mass effects on micro-bial community assembly in groundwater during riverbank filtration. Water Res. 2021, 189, 116631. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gomez-Velez, J.D.; Wilson, J.L. Dynamic coevolution of baseflow and multiscale groundwater flow system during prolonged droughts. J. Hydrol. 2022, 609, 127657. [Google Scholar] [CrossRef]
- Retter, A.; Karwautz, C.; Griebler, C. Groundwater microbial communities in times of climate change. Curr. Issues Mol. Biol. 2020, 41, 509–538. [Google Scholar] [PubMed]
- Rivera, A. Canada’s Groundwater Resources; Fitzhenry & Whiteside: Toronto, ON, Canada, 2014. [Google Scholar]
- Zhang, Y.; Dekas, A.E.; Hawkins, A.J.; Parada, A.E.; Gorbatenko, O.; Li, K.; Horne, R.N. Microbial community composition in deep-subsurface reservoir fluids reveals natural interwell connectivity. Water Resour. Res. 2019, 56, e2019WR025916. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.-W.; Lee, Y.-W.; Kim, G. Large submarine groundwater discharge and benthic eutrophication in Bangdu Bay on volcanic Jeju Island, Korea. Limnol. Oceanogr. 2005, 50, 1393–1403. [Google Scholar] [CrossRef]
- Lee, E.; Shin, D.; Hyun, S.P.; Ko, K.-S.; Moon, H.S.; Koh, D.-C.; Ha, K.; Kim, B.-Y. Periodic change in coastal microbial community structure associated with submarine groundwater discharge and tidal fluctuation. Limnol. Oceanogr. 2017, 62, 437–451. [Google Scholar] [CrossRef]
- Adyasari, D.; Hassenrück, C.; Montiel, D.; Dimova, N. Microbial community composition across a coastal hydrological system affected by submarine groundwater discharge (SGD). PLoS ONE 2020, 15, e0235235. [Google Scholar] [CrossRef]
- Glamoclija, M.; Ramirez, S.; Sirisena, K.; Widanagamage, I. Subsurface microbial ecology at sediment-groundwater interface in sulfate-rich Playa; White Sands National Monument, New Mexico. Front. Microbiol. 2019, 10, 2595. [Google Scholar] [CrossRef] [Green Version]
- Korbel, K.L.; Rutlidge, H.; Hose, G.C.; Eberhard, S.M.; Andersen, M.S. Dynamics of microbiotic patterns reveal surface water groundwater interactions in intermittent and perennial streams. Sci. Total Environ. 2022, 811, 152380. [Google Scholar] [CrossRef]
- Le Projet D’acquisition De Connaissances Sur Les Eaux Souterraines Dans La Région Des Laurentides Et De La MRC Les Moulins. 2022. Available online: https://archipel.uqam.ca/15786/ (accessed on 21 June 2023).
- Groult, B.; Bredin, P.; Lazar, C.S. Ecological processes differ in community assembly of Archaea, Bacteria and Eukaryotes in a biogeographical survey of groundwater habitats in the Quebec region. Environ. Microbiol. 2022, 24, 5898–5910. [Google Scholar] [CrossRef]
- Gantner, S.; Andersson, A.F.; Alonso-Sáez, L.; Bertilsson, S. Novel primers for 16S rRNA-based archaeal community analyses in environmental samples. J. Microbiol. Methods 2011, 84, 12–18. [Google Scholar] [CrossRef]
- Stahl, D.A.; Amann, R.I. Development and Application of Nucleic Acid Probes. In Nucleic Acid Techniques in Bacterial Systematics; John Wiley & Sons: Hoboken, NJ, USA, 1991; pp. 205–248. [Google Scholar]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; De Rijk, P.; Wuyts, J.; Winkelmans, T.; De Wachter, R. The European small subunit ribosomal RNA database. Nucleic Acids Res. 2000, 28, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Gast, R.J.; Dennett, M.R.; Caron, D.A. Characterization of protistan assemblages in the Ross Sea, Antarctica, by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 2004, 70, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D. Reintroducing mothur: 10 Years Later. Appl. Environ. Microbiol. 2020, 86, e02343-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene data-base project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Zhou, Z.; Pan, J.; Wang, F.; Gu, J.-D.; Li, M. Bathyarchaeota: Globally distributed metabolic generalists in anoxic environments. FEMS Microbiol. Rev. 2018, 42, 639–655. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Castelle, C.J.; Probst, A.J.; Zhou, Z.; Pan, J.; Liu, Y.; Banfield, J.F.; Gu, J.-D. Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 2018, 6, 102. [Google Scholar] [CrossRef]
- Pereira, M.B.; Wallroth, M.; Jonsson, V.; Kristiansson, E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genom. 2018, 19, 274. [Google Scholar] [CrossRef] [Green Version]
- R Core Team, R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Shenhav, L.; Thompson, M.; Joseph, T.A.; Briscoe, L.; Furman, O.; Bogumil, D.; Mizrahi, I.; Peer, I.; Halperin, E. FEAST: Fast expectation-maximization for microbial source tracking. Nat. Methods 2019, 16, 627–632. [Google Scholar] [CrossRef]
- Capo, E.; Ninnes, S.; Domaizon, I.; Bertilsson, S.; Bigler, C.; Wang, X.-R.; Bindler, R.; Rydberg, J. Landscape setting drives the microbial eukaryotic community structure in four Swedish mountain lakes over the Holocene. Microorganisms 2021, 9, 355. [Google Scholar] [CrossRef] [PubMed]
- Potvin, M.; Rautio, M.; Lovejoy, C. Freshwater microbial eukaryotic core communities, open-water and under-ice specialists in southern Victoria Island lakes (Ekaluktutiak, NU, Canada). Front. Microbiol. 2022, 12, 786094. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, H.; Zeng, Y.; Ding, H.; Wang, B.; Li, Y.; Ji, Z.; Bi, Y.; Luo, W. Diversity and assembly processes of microbial eukaryotic communities in Fildes Peninsula Lakes (West Antarctica). Biogeosciences 2022, 19, 4639–4654. [Google Scholar] [CrossRef]
- Inkinen, J.; Jayaprakash, B.; Siponen, S.; Hokajärvi, A.-M.; Pursiainen, A.; Ikonen, J.; Ryzhikov, I.; Täubel, M.; Kauppinen, A.; Paananen, J.; et al. Active eukaryotes in drinking water distribution systems of ground and surface waterworks. Microbiome 2019, 7, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, M.; Geeskink, P.; Yan, L.; Lehmann, R.; Totsche, K.-U.; Küsel, K. Complex food webs coincide with high genetic potential for chemolithoautotrophy in fractured bedrock groundwater. Water Res. 2020, 170, 115306. [Google Scholar] [CrossRef]
- Brett, M.T.; Bunn, S.E.; Chandra, S.; Galloway, A.W.E.; Guo, F.; Kainz, M.J.; Kankaala, P.; Lau, D.C.P.; Moulton, T.P.; Power, M.E.; et al. How important are terrestrial organic carbon inputs for secondary production in freshwater ecosystems? Freshw. Biol. 2017, 62, 833–853. [Google Scholar] [CrossRef]
- Fitch, A.; Orland, C.; Willer, D.; Smilson, E.J.S.; Tanentzap, A.J. Feasting on terrestrial organic matter: Dining in a dark lake changes microbial decomposition. Glob. Chang. Biol. 2018, 24, 5110–5122. [Google Scholar] [CrossRef] [Green Version]
- Pernthaler, J. Predation on prokaryotes in the water column and its ecological implications. Nat. Rev. Microbiol. 2005, 3, 537–546. [Google Scholar] [CrossRef]
- Jousset, A. Ecological and evolutive implications of bacterial defences against predators. Environ. Microbiol. 2012, 14, 1830–1843. [Google Scholar] [CrossRef]
- Akob, D.M.; Küsel, K. Where microorganisms meet rocks in the Earth’s Critical Zone. Biogeosciences 2011, 8, 3531–3543. [Google Scholar] [CrossRef] [Green Version]
- Bay, S.K.; Waite, D.W.; Dong, X.; Gillor, O.; Chown, S.L.; Hugenholtz, P.; Greening, C. Chemosynthetic and photosynthetic bacteria contribute differentially to primary production across a steep desert aridity gradient. ISME J. 2021, 15, 3339–3356. [Google Scholar] [CrossRef]
- Griebler, C.; Lueders, T. Microbial biodiversity in groundwater ecosystems. Freshw. Biol. 2009, 54, 649–677. [Google Scholar] [CrossRef]
- Shang, Y.; Wu, X.; Wang, X.; Wei, Q.; Ma, S.; Sun, G.; Zhang, H.; Wang, L.; Dou, H.; Zhang, H. Factors affecting seasonal variation of microbial community structure in Hulun Lake, China. Sci. Total Environ. 2022, 805, 150294. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, A.; Purahong, W.; Lehmann, R.; Herrmann, M.; Küsel, K.; Totsche, K.-U.; Buscot, F.; Wubet, T. Superimposed pristine limestone aquifers with marked hydrochemical differences exhibit distinct fungal communities. Front. Microbiol. 2016, 7, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefèvre, E.; Roussel, B.; Amblard, C.; Sime-Ngando, T. The molecular diversity of freshwater picoeukaryotes reveals high occurrence of putative parasitoids in the plankton. PLoS ONE 2008, 3, e2324. [Google Scholar] [CrossRef]
- Delafont, V.; Rodier, M.-H.; Maisonneuve, E.; Cateau, E. Vermamoeba vermiformis: A Free-Living Amoeba of Interest. Microb. Ecol. 2018, 76, 991–1001. [Google Scholar] [CrossRef]
- Kulas, A.; Gulin, V.; Kepcija, R.M.; Zutinic, P.; Peric, M.S.; Orlic, S.; Kajan, K.; Stoeck, T.; Lentendu, G.; Canjevac, I.; et al. Ciliates (Alveolata, Ciliophora) as bioindicators of environmental pressure: A karstic case. Ecol. Indic 2021, 124, 107430. [Google Scholar] [CrossRef]
- Cozannet, M.; Borrel, G.; Roussel, E.; Moalic, Y.; Allioux, M.; Sanvoisin, A.; Toffin, L.; Alain, K. New insights into the ecology and physiology of Methanomassiliicoccales from terrestrial and aquatic environments. Microorganisms 2021, 9, 30. [Google Scholar] [CrossRef]
- Guerrero-Cruz, S.; Cremers, G.; van Alen, T.A.; Op den Camp, H.J.M.; Jetten, M.S.M.; Rasigraf, O.; Vaksmaa, A. Response of the anaerobic methanotroph “Candidatus Methanoperedens nitroreducens” to oxygen stress. Appl. Environ. Microbiol. 2018, 84, e01832-18. [Google Scholar] [CrossRef] [Green Version]
- Blainey, P.C.; Mosier, A.C.; Potanina, A.; Francis, C.A.; Quake, S.R. Genome of a low-salinity ammonia-oxidizing archaeon deter-mined by single-cell and metagenomic analysis. PLoS ONE 2011, 6, e16626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbold, C.W.; Lehtovirta-Morley, L.E.; Jung, M.-Y.; Jehmlich, N.; Hausman, B.; Han, P.; Loy, A.; Pester, M.; Sayavedra-Soto, L.A.; Rhee, S.-K.; et al. Ammonia-oxidising archaea living at low pH: Insights from comparative genomics. Environ. Microbiol. 2017, 19, 4939–4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellbye, B.L.; Bottomley, P.J.; Sayavedra-Soto, L.A. Nitrite-oxidizing bacterium Nitrobacter winogradskyi produces N-Acyl-Homoserine lactone autoinducers. Appl. Environ. Microbiol. 2015, 81, 5917–5926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, B.; Saito, M.A.; McIlvin, M.R.; Lucker, S.; Moran, D.M.; Lankiewicz, T.S.; Dupont, C.L.; Santoro, A.E. Metabolic versatility of the nitrite-oxidizing bacterium Nitrospira marina and its proteomic response to oxygen-limited conditions. ISME J. 2021, 15, 1025–1039. [Google Scholar] [CrossRef] [PubMed]
- Issotta, F.; Moya-Beltran, A.; Mena, C.; Covarrubias, P.C.; Thyssen, C.; Bellenberg, S.; Sand, W.; Quatrini, R.; Vera, M. Insights into the biology of acidophilic members of the Acidiferrobacteraceae family derived from comparative genomic analyses. Res. Microbiol. 2018, 169, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Fukui, M. Sulfuricella denitrificans gen. nov., sp. nov., a sulfur-oxidizing autotroph isolated from a freshwater lake. IJSEM 2010, 60, 2862–2866. [Google Scholar] [CrossRef] [Green Version]
- Hallbeck, L.; Karsten, D. Autotrophic and mixotrophic growth of Gallionella ferruginea. J. Gen. Microbiol. 1991, 137, 2657–2661. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Liu, Y.; Xu, W.; Pan, J.; Luo, Z.-H.; Li, M. Genome- and community-level interaction insights into carbon utilization and element cycling functions of Hydrothermarchaeota in hydrothermal sediment. mSystems 2020, 5, e00795-19. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, M.; Rusznyak, A.; Akob, D.M.; Schukze, I.; Opitz, S.; Totsche, K.-U.; Küsel, K. Large fractions of CO2-fixing microorganisms in pristine limestone aquifers appear to be involved in the oxidation of reduced sulfur and nitrogen compounds. Appl. Environ. Microbiol. 2015, 81, 2384–2394. [Google Scholar] [CrossRef] [Green Version]
- Fabisch, M.; Beulig, F.; Akob, D.M.; Küsel, K. Surprising abundance of Gallionella-related iron-oxidizers in creek sediments at pH 4.4 or at high heavy metal concentrations. Front. Microbiol. 2013, 4, 390. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Jiang, Y.; Liu, Q.; Wang, R.; Liu, X.; Liu, B. Enzymatic and regulatory properties of the trehalose-6-phosphate synthase from the thermoacidophilic archaeon Thermoplasma acidophilum. Biochimie 2014, 101, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Graml, W.; Santos-Martinez, M.-L.; Koretke, C.; Volker, C.; Mewes, H.W.; Frishman, D.; Sotcker, S.; Lupas, A.N.; Baumeister, W. The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature 2000, 407, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoki, T.; Otsuka, Y.; Ikeda, S.; Masai, E.; Kajita, S.; Katayama, Y. Tetrahydrofolate-dependent vanillate and syringate O-demethylation links tightly to one-carbon metabolic pathway associated with amino acid synthesis and DNA methylation in the lignin metabolism of Sphingomonas paucimobilis SYK-6. J. Wood. Sci. 2002, 48, 434–439. [Google Scholar] [CrossRef]
- Das, S.; Barooah, M. Characterization of siderophore producing arsenic-resistant Staphylococcus sp. Strain TA6 isolated from contaminated groundwater of Jorhat, Assam and its possible role in arsenic geocycle. BMC Microbiol. 2018, 18, 104. [Google Scholar] [CrossRef] [Green Version]
- Hoef-Emden, K.; Melkonian, M. Revision of the genus Cryptomonas (Cryptophyceae): A combination of molecular phylogeny and morphology provides in-sights into a long-hidden dimorphism. Protist 2003, 154, 371–409. [Google Scholar] [CrossRef]
- Santoferrara, L.F.; Alder, V.V.; McManus, G.B. Phylogeny, classification and diversity of Choreotrichia and Oligotrichia (Ciliophora, Spirotrichea). Mol. Phylogenet. Evol. 2017, 112, 12–22. [Google Scholar] [CrossRef]
- DeLong, J.P.; Van Etten, J.; Al-Almeeli, A.; Agarkova, I.V.; Dunigan, D.D. The consumption of viruses returns energy to food chains. Ecology 2023, 120, e2215000120. [Google Scholar] [CrossRef]
- Wallace, R.L.; Ott, D.W.; Stiles, S.L.; Oldham-Ott, C.K. Bed and Breakfast: The parasitic life of Proales werneckii (Ploimida: Proalidae) within the alga Vaucheria (Xanthophyceae: Vaucheriales). Hydrobiologia 2001, 446, 129–137. [Google Scholar] [CrossRef]
- Grossart, H.-P.; Frindte, K.; Dziallas, C.; Eckert, W.; Tang, K.W. Microbial methane production in oxygenated water column of an oligotrophic lake. Proc. Natl. Acad. Sci. USA 2011, 108, 19657–19661. [Google Scholar] [CrossRef] [Green Version]
- Bogard, M.J.; del Giorgio, P.A.; Boutet, L.; Chaves, M.C.G.; Prairie, Y.T.; Merante, A.; Derry, A.M. Oxic water column methanogenesis as a major com-ponent of aquatic CH4 fluxes. Nat. Commun. 2014, 5, 6350. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Hu, C.; Wang, Y.; Wu, J.; Hing, Y. Biogeographic pattern of methanogenic community in surface water along the Yangtze River. Geomicrobiol. J. 2021, 38, 588–597. [Google Scholar] [CrossRef]
- Pan, J.; Zhou, Z.; Beja, O.; Cai, M.; Yang, Y.; Liu, Y.; Gu, J.-D.; Li, M. Genomic and transcriptomic evidence of light-sensing, porphyrin biosynthesis, Calvin-Benson-Bassham cycle, and urea production in Bathyarchaeota. Microbiome 2020, 8, 43. [Google Scholar] [CrossRef]
- Liu, J.; Fu, B.; Yang, H.; Zhao, M.; He, B.; Zhang, X.-H. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: The potential impact of hypoxia and nutrients. Front. Microbiol. 2015, 6, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.W.; Lang, E.; Brandt, U.; Lundsdorf, H.L.; Wu, Q.L.; Stackebrandt, E. Polynucleobacter cosmopolitanus sp. nov., free-living planktonic bacte-ria inhabiting freshwater lakes and rivers. IJSEM 2010, 60, 166–173. [Google Scholar] [PubMed]
- Hahn, M.W.; Lang, E.; Brandt, U.; Sproer, C. Polynucleobacter acidiphobus sp. nov., a representative of an abundant group of planktonic freshwater bacteria. IJSEM 2011, 61, 788–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasalicky, V.; Jezbera, J.; Simek, K.; Hahn, M.W. Limnohabitans planktonicus sp. nov. and Limnohabitans parvus sp. nov., planktonic betaproteobacteria isolated from a freshwater reservoir, and emended description of the genus Limnohabitans. IJSEM 2010, 60, 2710–2714. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.W.; Kasalicky, V.; Jezbra, J.; Brandt, U.; Jezberova, J.; Simek, K. Limnohabitans curvus gen. nov., sp. nov., a planktonic bacterium isolated from a freshwater lake. IJSEM 2010, 60, 1358–1365. [Google Scholar] [CrossRef]
- Spring, S.; Kampfer, P.; Schleifer, K.H. Limnobacter thiooxidans gen. nov., sp. nov., a novel thiosulfate-oxidizing bacterium isolated from freshwater lake sediment. IJSEM 2001, 51, 1463–1470. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.W. Description of seven candidate species affiliated with the phylum Actino-bacteria, representing planktonic freshwater bacteria. Int. J. Syst. Evol. Microbiol. 2009, 59, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.W.; Pitt, A.; Schmidt, J.; Koll, U.; Wolf, J.; Whitman, W.B.; Bodelier, P.L.E.; Neumann-Schaal, M. Zwartia hollandica gen. nov., sp. nov., Jezberella montanilacus gen. nov., sp. nov. and Sheuella amnicola gen. nov., comb. nov., representing the environmental GKS98 (betIII) cluster. Int. J. Syst. Evol. Microbiol. 2022, 72, 005513. [Google Scholar] [CrossRef]
- Qu, J.-H.; Zhang, L.-J.; Fu, Y.-H.; Li, X.-D.; Li, H.-F.; Tian, H.-L. A novel genus of the class Actinobacteria, Longivirga aurantiaca gen. nov., sp. nov., isolated from lake sediment. Int. J. Syst. Evol. Microbiol. 2018, 68, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Chern, L.-L.; Stackebrandt, E.; Lee, S.-F.; Lee, F.-L.; Chen, J.-K.; Fu, H.-M. Chitinibacter tainanensis gen. nov., sp. nov., a chitin-degrading aerobe from soil in Taiwan. IJSEM 2004, 54, 1387–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, A. The Family Xanthobacteraceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin, Heidelberg, 2014. [Google Scholar] [CrossRef]
- Willems, A. The Family Comamonadaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin, Heidelberg, 2014. [Google Scholar] [CrossRef]
- Chen, L.X.; Mendez-Garcia, C.; Dombrowski, N.; Servin-Garciduenas, L.E.; Eloa-Fafrosh, E.A.; Fang, B.-Z.; Luo, Z.-H.; Tan, S.; Zhi, X.-Y.; Hua, Z.-S.; et al. Metabolic versatility of small archaea Micrarchaeota and Parvarchaeota. ISME J. 2018, 12, 756–775. [Google Scholar] [CrossRef] [PubMed]
- McConnaughey, M. Physical Chemical Properties of Fungi. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Dodds, W.K.; Whiles, M.R. Chapter 9—Microbes and Plants. In Aquatic Ecology, Freshwater Ecology, 2nd ed.; Dodds, W.K., Whiles, M.R., Eds.; Academic Press: Cambridge, MA, USA, 2010; pp. 185–219. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Df | SumOfSqs | R2 | F | Pr (>F) | |
|---|---|---|---|---|---|
| ARCHAEA | |||||
| Habitat | 1 | 0.9035 | 0.04216 | 1.8927 | 0.001 |
| Residual | 43 | 20.5271 | 0.95784 | ||
| Total | 44 | 21.4306 | 1.00000 | ||
| BACTERIA | |||||
| Habitat | 1 | 1.7682 | 0.06323 | 3.8472 | 0.001 |
| Residual | 57 | 26.1979 | 0.93677 | ||
| Total | 58 | 27.9662 | 1.00000 | ||
| EUKARYOTE | |||||
| Habitat | 1 | 1.5954 | 0.06515 | 3.5542 | 0.001 |
| Residual | 51 | 22.8929 | 0.93485 | ||
| Total | 52 | 24.4883 | 1.0000 |
| Df | SumOfSqs | F | Pr (>F) | |
|---|---|---|---|---|
| ARCHAEA | ||||
| DOC | 1 | 0.5443 | 1.1176 | 0.004 |
| DIC | 1 | 0.6213 | 1.2758 | 0.001 |
| Residuals | 42 | 20.4538 | ||
| BACTERIA | ||||
| DOC | 1 | 0.6047 | 1.2629 | 0.006 |
| DIC | 1 | 0.8119 | 1.6956 | 0.001 |
| Residuals | 56 | 26.8145 | ||
| EUKARYOTE | ||||
| DOC | 1 | 0.6600 | 1.4583 | 0.007 |
| DIC | 1 | 0.9515 | 2.1026 | 0.001 |
| Residuals | 50 | 22.6271 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groult, B.; St-Jean, V.; Lazar, C.S. Linking Groundwater to Surface Discharge Ecosystems: Archaeal, Bacterial, and Eukaryotic Community Diversity and Structure in Quebec (Canada). Microorganisms 2023, 11, 1674. https://doi.org/10.3390/microorganisms11071674
Groult B, St-Jean V, Lazar CS. Linking Groundwater to Surface Discharge Ecosystems: Archaeal, Bacterial, and Eukaryotic Community Diversity and Structure in Quebec (Canada). Microorganisms. 2023; 11(7):1674. https://doi.org/10.3390/microorganisms11071674
Chicago/Turabian StyleGroult, Benjamin, Vicky St-Jean, and Cassandre Sara Lazar. 2023. "Linking Groundwater to Surface Discharge Ecosystems: Archaeal, Bacterial, and Eukaryotic Community Diversity and Structure in Quebec (Canada)" Microorganisms 11, no. 7: 1674. https://doi.org/10.3390/microorganisms11071674