
Isolation and Characterization of the Acadevirus Members BigMira and MidiMira Infecting a Highly Pathogenic Proteus mirabilis Strain

 , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage Isolation, Propagation, and Purification
2.2. Biological Features
2.2.1. Lysis Plaque Measurements
2.2.2. Transmission Electron Microscopy (TEM)
2.2.3. Host Range
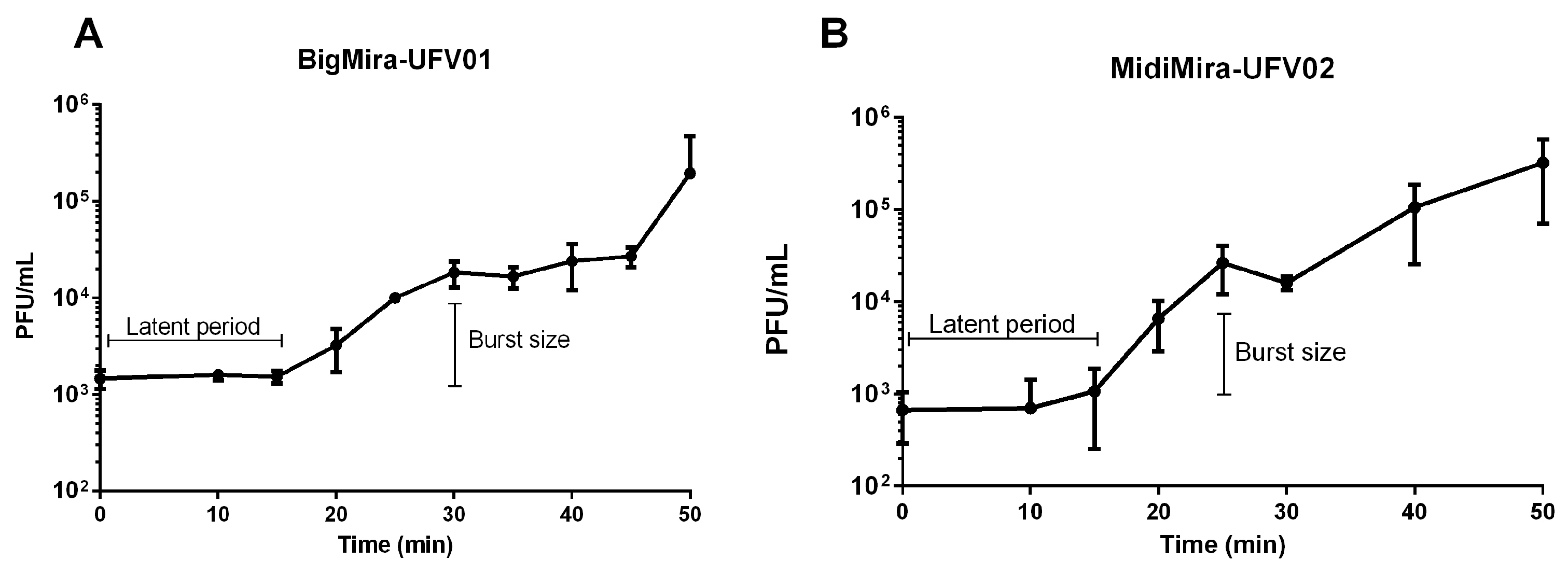
2.2.4. One-Step Growth Curve
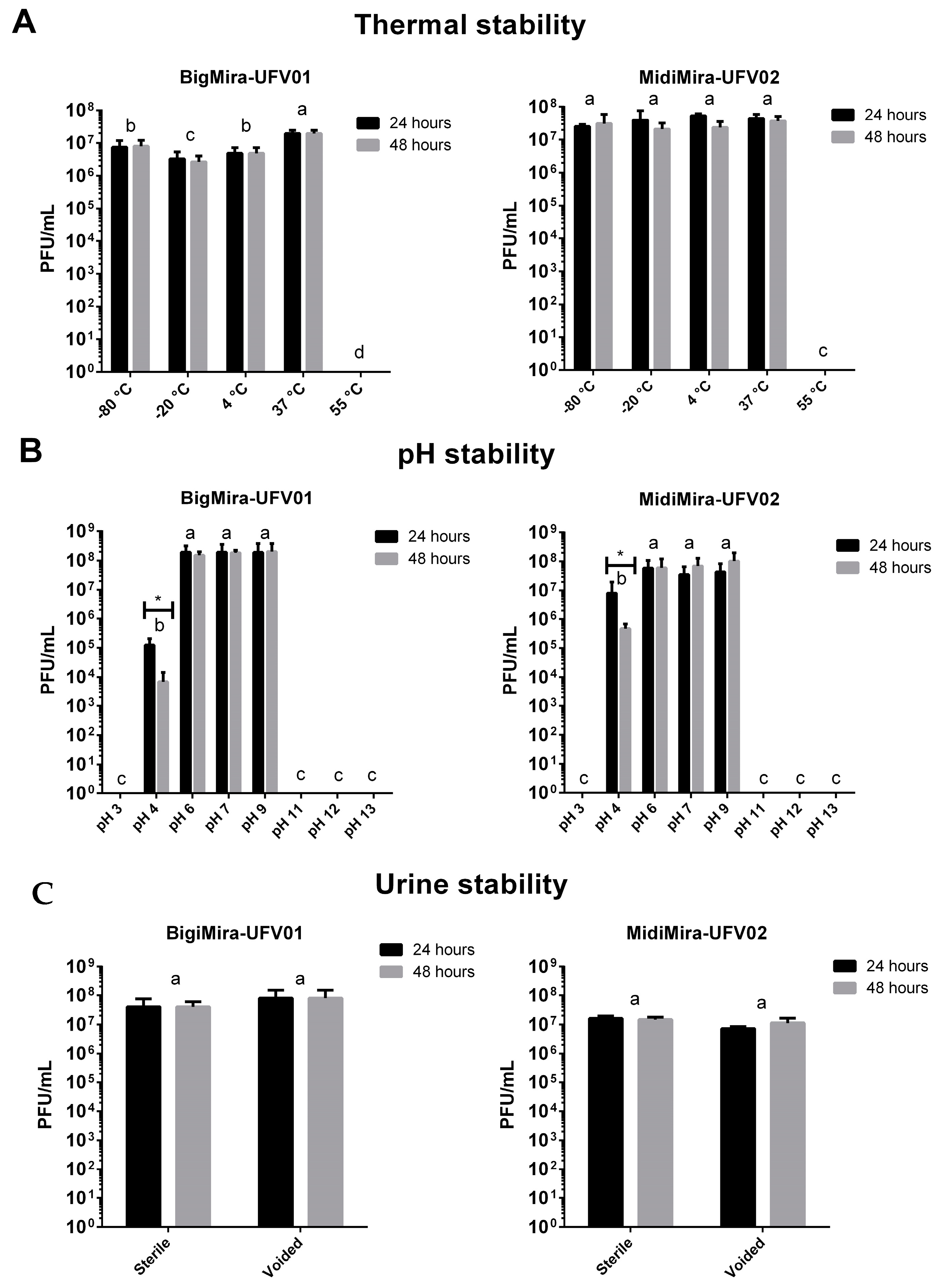
2.2.5. Phage Stability
2.3. Genome Analysis
2.3.1. DNA Extraction and Sequencing
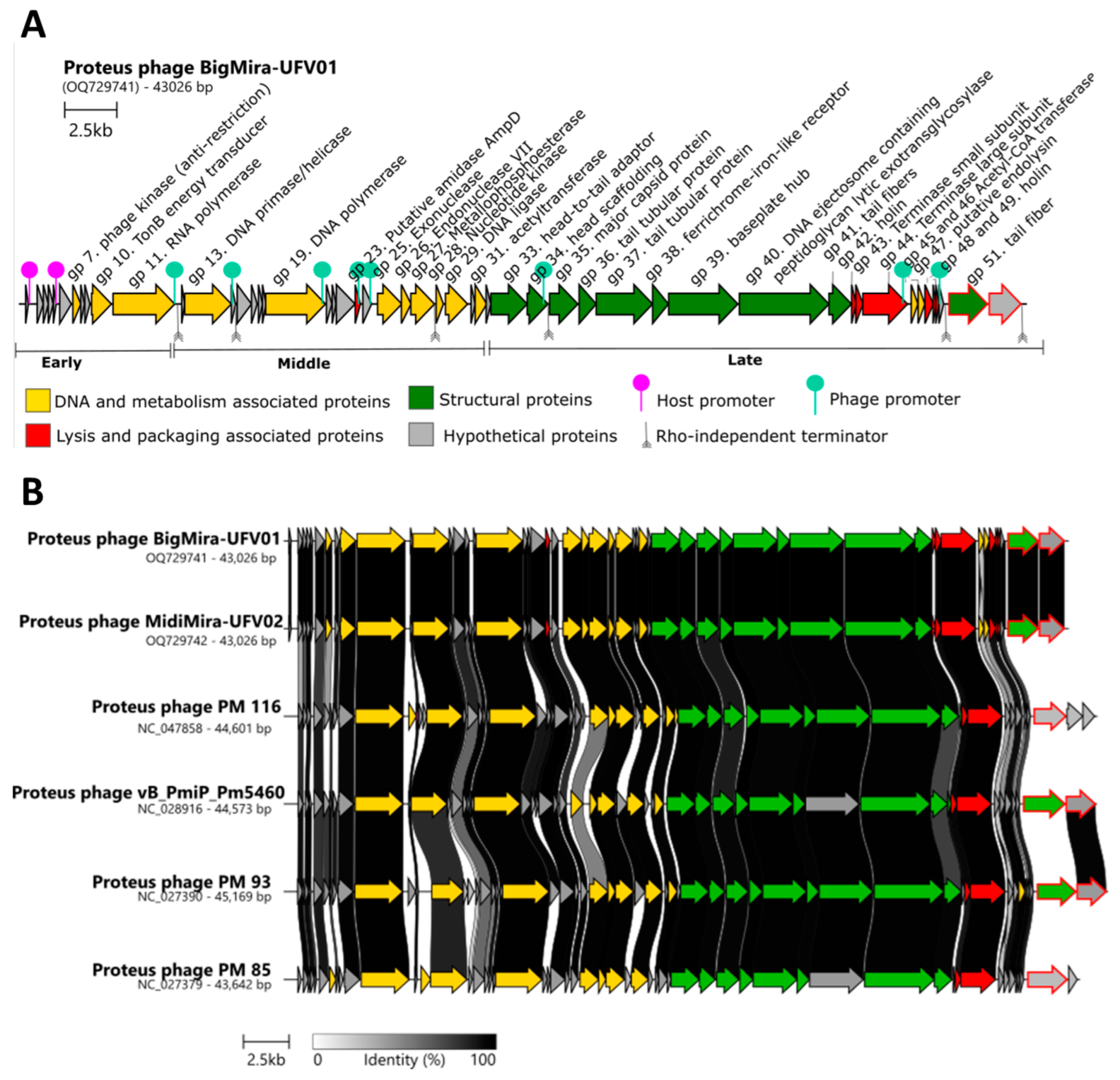
2.3.2. Assembly and Annotation
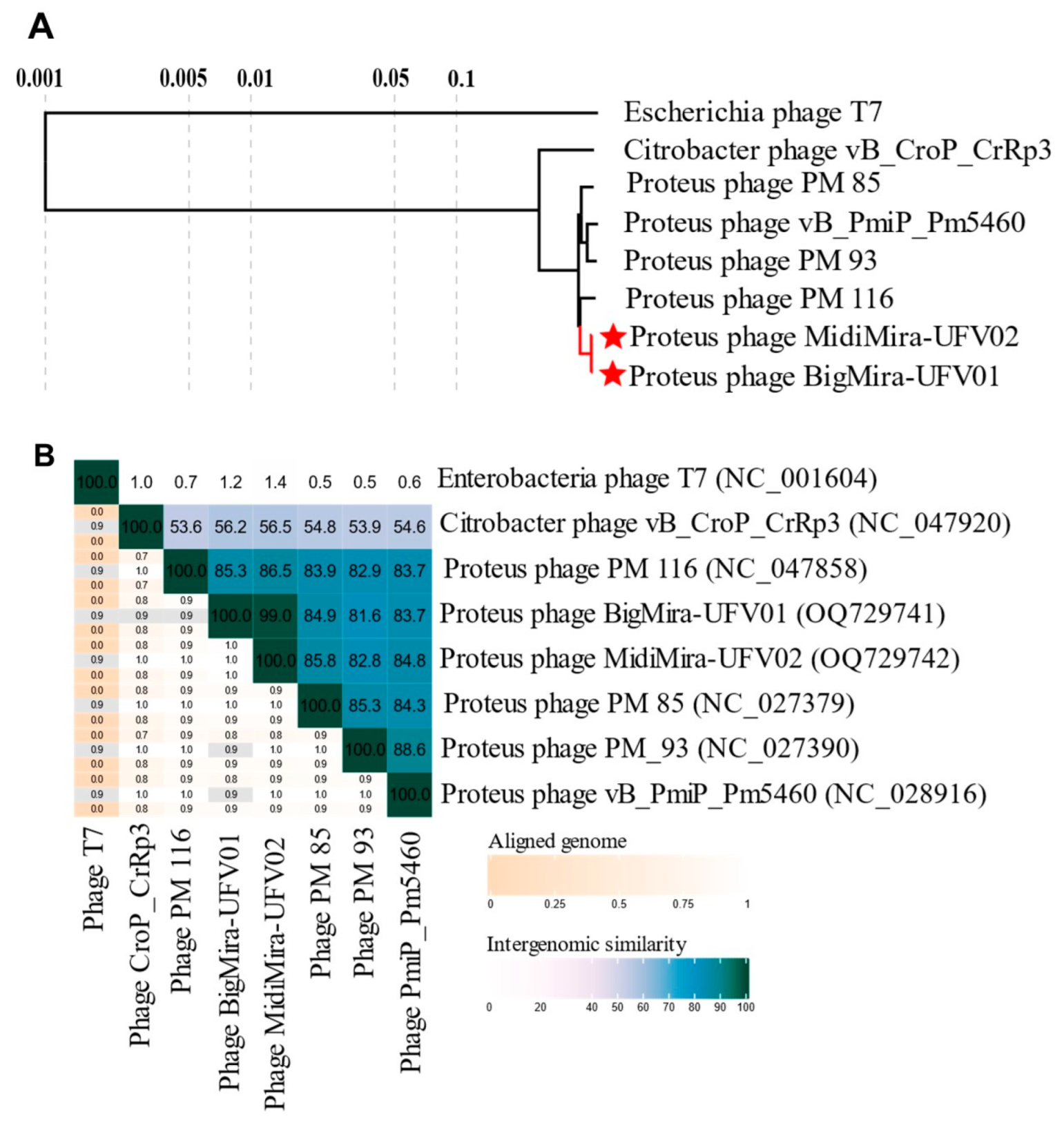
2.3.3. Genomic and Phylogenetic Analysis
2.3.4. Putative Depolymerase Enzyme Search and Tertiary Structure Prediction
2.4. Proteus mirabilis Clinical Strains
2.4.1. Bacterial Strains
2.4.2. Proteus mirabilis DNA Extraction and Sequencing
2.4.3. Genome Assembly and Annotation
2.4.4. Bioinformatics Analysis
3. Results
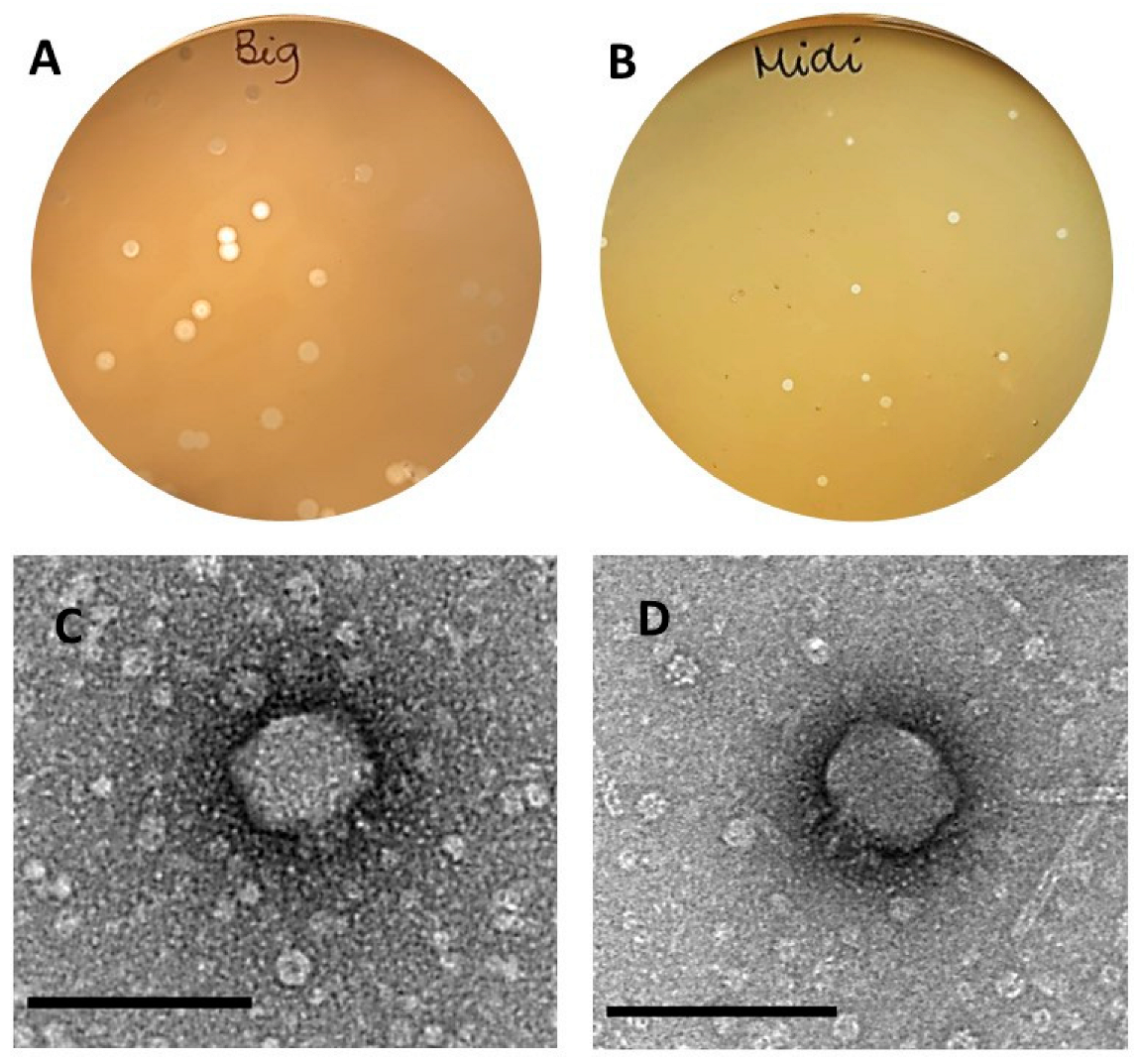
3.1. Phage Isolation
3.2. Biological Features
3.3. Genomic Features
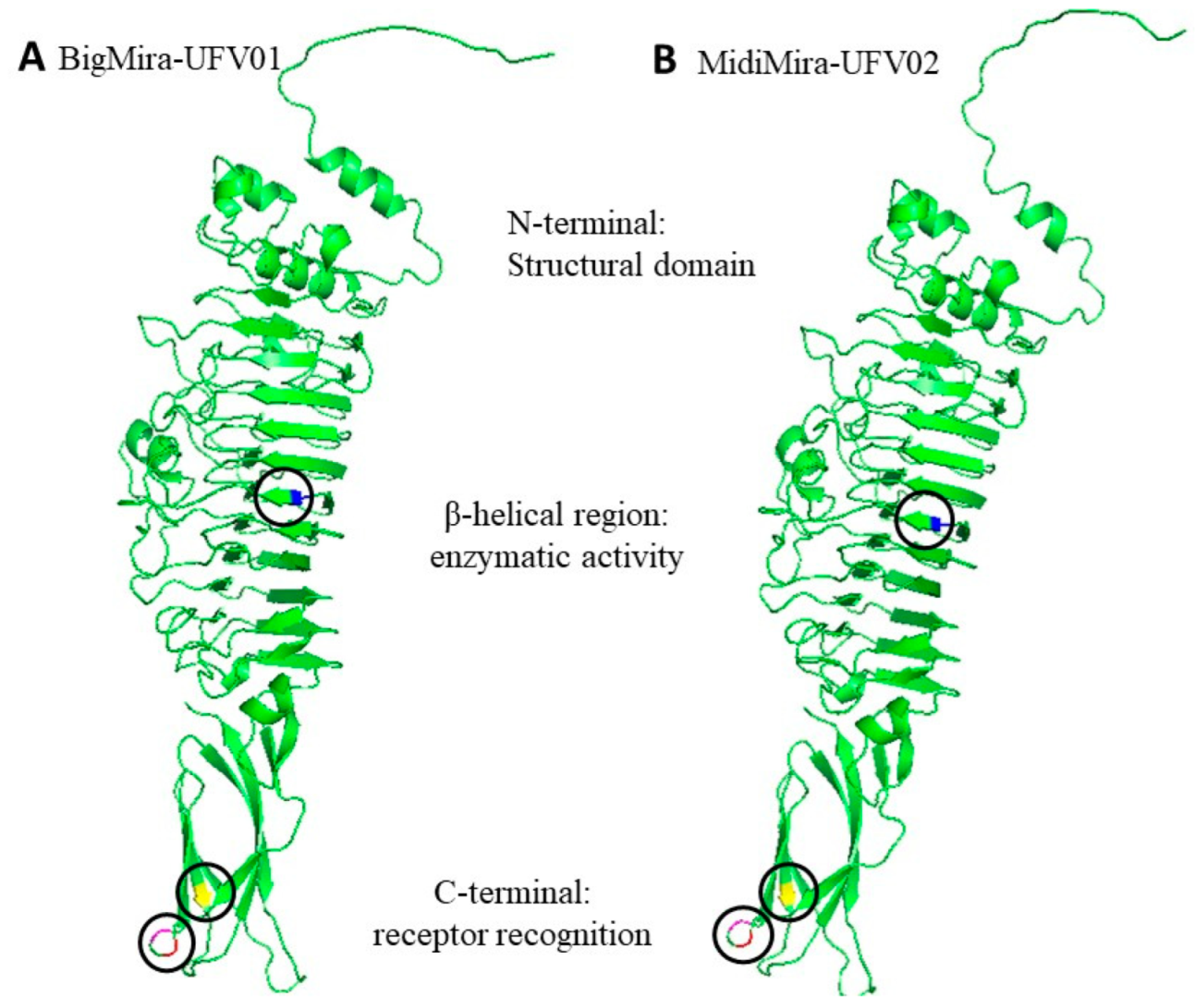
3.4. Putative Depolymerase-Encoding Domain Prediction
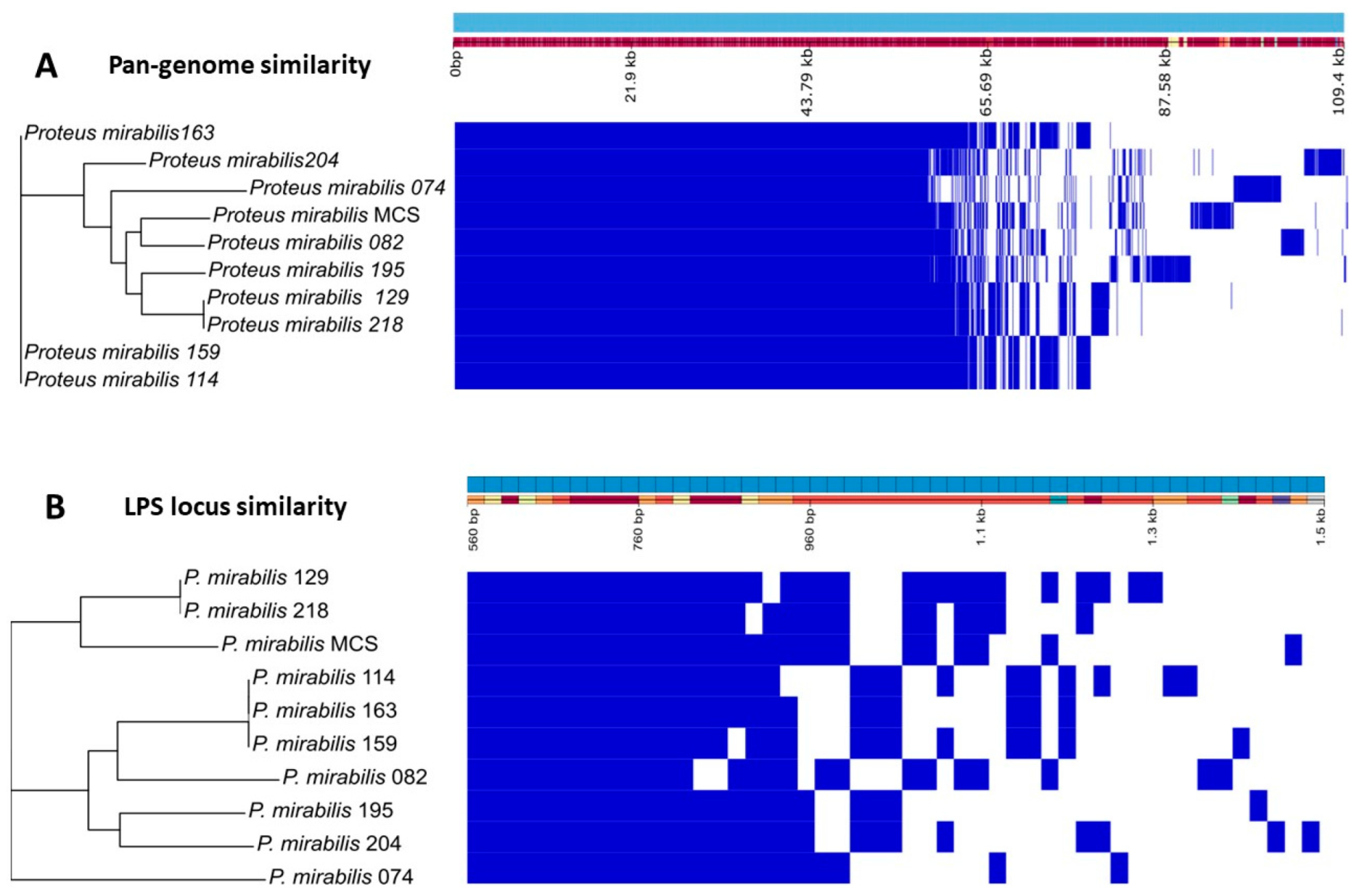
3.5. Proteus mirabilis Clinical Strain Genomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yuan, F.; Huang, Z.; Yang, T.; Wang, G.; Li, P.; Yang, B.; Li, J. Pathogenesis of Proteus mirabilis in Catheter-Associated Urinary Tract Infections. Urol. Int. 2021, 105, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Werneburg, G.T. Catheter-Associated Urinary Tract Infections: Current Challenges and Future Prospects. Res. Rep. Urol. 2022, 14, 109–133. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, L.E. Catheter associated urinary tract infections. Antimicrob. Resist. Infect. Control 2014, 3, 23. [Google Scholar] [CrossRef]
- Girlich, D.; Bonnin, R.A.; Dortet, L.; Naas, T. Genetics of Acquired Antibiotic Resistance Genes in Proteus spp. Front. Microbiol. 2020, 11, 256. [Google Scholar] [CrossRef]
- Li, Z.; Peng, C.; Zhang, G.; Shen, Y.; Zhang, Y.; Liu, C.; Liu, M.; Wang, F. Prevalence and characteristics of multidrug-resistant Proteus mirabilis from broiler farms in Shandong Province, China. Poult. Sci. 2022, 101, 101710. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, C.E.; Mobley, H.L.T.; Pearson, M.M. Pathogenesis of Proteus mirabilis Infection. EcoSal Plus 2018, 8, 10-1128. [Google Scholar] [CrossRef]
- Mirzaei, A.; Wagemans, J.; Nasr Esfahani, B.; Lavigne, R.; Moghim, S. A Phage Cocktail To Control Surface Colonization by Proteus mirabilis in Catheter-Associated Urinary Tract Infections. Microbiol. Spectr. 2022, 10, e0209222. [Google Scholar] [CrossRef] [PubMed]
- Fruciano, E.; Bourne, S. Phage as an antimicrobial agent: D’Herelle’s heretical theories and their role in the decline of phage prophylaxis in the West. Can. J. Infect. Dis. Med. Microbiol. 2007, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M.; Strauch, E. Phage therapy: Facts and fiction. Int. J. Med. Microbiol. 2006, 296, 5–14. [Google Scholar] [CrossRef]
- Doss, J.; Culbertson, K.; Hahn, D.; Camacho, J.; Barekzi, N. A review of phage therapy against bacterial pathogens of aquatic and terrestrial organisms. Viruses 2017, 9, 50. [Google Scholar] [CrossRef]
- Weber-d, B.; Majewska, J.; Borysowski, J.; Krystyna, D. Phage Therapy: Combating Infections with Potential for Evolving from Merely a Treatment for Complications to Targeting Diseases. Front. Microbiol. 2016, 7, 1515. [Google Scholar] [CrossRef]
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 278–283. [Google Scholar] [CrossRef]
- Razzaque, M.S. Commentary: Microbial Resistance Movements: An Overview of Global Public Health Threats Posed by Antimicrobial Resistance, and How Best to Counter. Front. Public Health 2021, 8, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention COVID-19 U.S. Impact on Antimicrobial Resistance; Department of Health and Human Services, CDC: Atlanta, GA, USA, 2022; pp. 1–44.
- Pan American Health Organization. Antimicrobial Resistance, Fueled by the COVID-19 Pandemic; Pan American Health Organization: Washington, DC, USA, 2022; pp. 1–14. [Google Scholar]
- Cahan, E. As Superbugs Flourish, Bacteriophage Therapy Recaptures Researchers’ Interest. J. Am. Med. Assoc. 2023, 329, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Langford, B.J.; Soucy, J.P.R.; Leung, V.; So, M.; Kwan, A.T.H.; Portnoff, J.S.; Bertagnolio, S.; Raybardhan, S.; MacFadden, D.R.; Daneman, N. Antibiotic resistance associated with the COVID-19 pandemic: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2023, 29, 302–309. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Ferry, T.; Kolenda, C.; Briot, T.; Souche, A.; Lustig, S.; Josse, J.; Batailler, C.; Pirot, F.; Medina, M.; Leboucher, G.; et al. Past and future of phage therapy and phage-derived proteins in patients with bone and joint infection. Viruses 2021, 13, 2414. [Google Scholar] [CrossRef]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162. [Google Scholar] [CrossRef]
- Pires, D.P.; Costa, A.R.; Pinto, G.; Meneses, L.; Azeredo, J. Current challenges and future opportunities of phage therapy. FEMS Microbiol. Rev. 2020, 44, 684–700. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef]
- Manohar, P.; Tamhankar, A.J.; Lundborg, C.S.; Nachimuthu, R. Therapeutic characterization and efficacy of bacteriophage cocktails infecting Escherichia coli, klebsiella pneumoniae, and enterobacter species. Front. Microbiol. 2019, 10, 574. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, R.; Díez-Martínez, R.; Domingo-Calap, P.; García, P.; Gutiérrez, D.; Muniesa, M.; Ruiz-Ruigómez, M.; Sanjuán, R.; Tomás, M.; Tormo-Mas, M.Á.; et al. Essential Topics for the Regulatory Consideration of Phages as Clinically Valuable Therapeutic Agents: A Perspective from Spain. Microorganisms 2022, 10, 717. [Google Scholar] [CrossRef] [PubMed]
- Naureen, Z.; Malacarne, D.; Anpilogov, K.; Dautaj, A.; Camilleri, G.; Cecchin, S.; Bressan, S.; Casadei, A.; Albion, E.; Sorrentino, E.; et al. Comparison between American and European legislation in the therapeutical and alimentary bacteriophage usage. Acta Biomed. 2020, 91, e2020023. [Google Scholar] [CrossRef] [PubMed]
- Verbeken, G.; Pirnay, J.P. European regulatory aspects of phage therapy: Magistral phage preparations. Curr. Opin. Virol. 2022, 52, 24–29. [Google Scholar] [CrossRef]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2018, 19, 35–45. [Google Scholar] [CrossRef]
- Onsea, J.; Uyttebroek, S.; Chen, B.; Wagemans, J.; Lood, C.; Van Gerven, L.; Spriet, I.; Devolder, D.; Debaveye, Y.; Depypere, M.; et al. Bacteriophage therapy for difficult-to-treat infections: The implementation of a multidisciplinary phage task force (the phageforce study protocol). Viruses 2021, 13, 1543. [Google Scholar] [CrossRef]
- Onsea, J.; Soentjens, P.; Djebara, S.; Merabishvili, M.; Depypere, M.; Spriet, I.; De Munter, P.; Debaveye, Y.; Nijs, S.; Vanderschot, P.; et al. Bacteriophage application for difficult-to-treat musculoskeletal infections: Development of a standardized multidisciplinary treatment protocol. Viruses 2019, 11, 891. [Google Scholar] [CrossRef]
- Uyttebroek, S.; Chen, B.; Onsea, J.; Ruythooren, F.; Debaveye, Y.; Devolder, D.; Spriet, I.; Depypere, M.; Wagemans, J.; Lavigne, R.; et al. Safety and efficacy of phage therapy in difficult-to-treat infections: A systematic review. Lancet Infect. Dis. 2022, 22, e208–e220. [Google Scholar] [CrossRef]
- Liu, D.; Van Belleghem, J.D.; de Vries, C.R.; Burgener, E.; Chen, Q.; Manasherob, R.; Aronson, J.R.; Amanatullah, D.F.; Tamma, P.D.; Suh, G.A. The safety and toxicity of phage therapy: A review of animal and clinical studies. Viruses 2021, 13, 1268. [Google Scholar] [CrossRef]
- Twest, R.; Kropinski, A.M. Bacteriophages: Methods and Protocols, Volume 1; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Leicester, UK, 2009; ISBN 9781588296825. [Google Scholar]
- Yamamoto, K.R.; Alberts, B.M.; Benzinger, R.; Lawhorne, L.; Treiber, G. Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification. Virology 1970, 40, 734–744. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Image to ImageJ: 25 years of image analysis. Nature methods. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Vallino, M.; Rossi, M.; Ottati, S.; Martino, G.; Galetto, L.; Marzachì, C.; Abbà, S. Bacteriophage-host association in the phytoplasma insect vector euscelidius variegatus. Pathogens 2021, 10, 612. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.D.R.; Veiga, P.; Cerca, N.; Kropinski, A.M.; Almeida, C.; Azeredo, J.; Sillankorva, S. Development of a phage cocktail to control Proteus mirabilis catheter-associated urinary tract infections. Front. Microbiol. 2016, 7, 1024. [Google Scholar] [CrossRef] [PubMed]
- Kot, W. Genome Sequencing of dsDNA-Containing Bacteriophages Directly from a Single Plaque. In Bacteriophages: Methods and Protocols, Volume 3; Al, M.R.J.C., Ed.; Springer Science: Berlin/Heidelberg, Germany; Business Media: Petaling Jaya, Malaysia, 2018; Volume 1681, pp. 179–184. ISBN 9781493973439. [Google Scholar]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Coppens, L.; Lavigne, R. SAPPHIRE: A neural network based classifier for σ70 promoter prediction in Pseudomonas. BMC Bioinform. 2020, 21, 415. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Naville, M.; Ghuillot-Gaudeffroy, A.; Marchais, A.; Gautheret, D. ARNold: A web tool for the prediction of rho-independent transcription terminators. RNA Biol. 2011, 8, 11–13. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.F.; Duarte, J.; Domingues, R.; Oliveira, H.; Dias, O. PhageDPO: Phage Depolymerase Finder. bioRxiv 2023. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, 244–248. [Google Scholar] [CrossRef]
- Alva, V.; Nam, S.Z.; Soding, J.; Lupas, A.N. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef]
- Morozova, V.; Kozlova, Y.; Shedko, E.; Babkin, I.; Kurilshikov, A.; Bokovaya, O.; Bardashova, A.; Yunusova, A.; Tikunov, A.; Tupikin, A.; et al. Isolation and characterization of a group of new Proteus bacteriophages. Arch. Virol. 2018, 163, 2189–2197. [Google Scholar] [CrossRef]
- Hung, E.; Darouiche, R.; Trautner, B. Proteus bacteriuria is associated with significant morbidity in spinal cord injury. Spinal Cord. NIH Public Access 2008, 23, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Wasfi, R.; Hamed, S.M.; Amer, M.A.; Fahmy, L.I. Proteus mirabilis Biofilm: Development and Therapeutic Strategies. Front. Cell. Infect. Microbiol. 2020, 10, 414. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, E.; Song, J.; Yang, L.; Wu, B. Complete Genome Sequence of a Novel T7-Like Bacteriophage from a Pasteurella multocida Capsular Type A Isolate. Curr. Microbiol. 2018, 75, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Elhalag, K.; Eldin, M.N.; Hussien, A.; Ahmad, A. Potential use of soilborne lytic podoviridae phage as a biocontrol agent against Ralstonia solanacearum. J. Basic Microbiol. 2018, 58, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tian, F.; Li, J.; Li, L.; Qiao, H.; Dong, Y.; Ma, F.; Zhu, S.; Tong, Y. Isolation and characterization of a podovirus infecting the opportunist pathogen Vibrio alginolyticus and Vibrio parahaemolyticus. Virus Res. 2021, 302, 198481. [Google Scholar] [CrossRef]
- Cole, A.W.; Tran, S.D.; Ellington, A.D. Heat adaptation of phage T7 under an extended genetic code. Virus Evol. 2021, 7, veab100. [Google Scholar] [CrossRef] [PubMed]
- Favor, A.H.; Llanos, C.D.; Youngblut, M.D.; Bardales, J.A. Optimizing bacteriophage engineering through an accelerated evolution platform. Sci. Rep. 2020, 10, 13981. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Dams, D.; Brøndsted, L.; Drulis-kawa, Z. Engineering of receptor-binding proteins in bacteriophages and phage tail-like bacteriocins. Biochem. Soc. Trans. 2019, 47, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Drulis-Kawa, Z.; Azeredo, J. Exploiting phage-derived carbohydrate depolymerases for combating infectious diseases. Trends Microbiol. 2022, 30, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, D.J. Amino Acids, Peptides, and Proteins; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128160619. [Google Scholar]
- Leveson-Gower, R.B.; Mayer, C.; Roelfes, G. The importance of catalytic promiscuity for enzyme design and evolution. Nat. Rev. Chem. 2019, 3, 687–705. [Google Scholar] [CrossRef]
- Squeglia, F.; Maciejewska, B.; Łątka, A.; Ruggiero, A.; Briers, Y.; Drulis-Kawa, Z.; Berisio, R. Structural and Functional Studies of a Klebsiella Phage Capsule Depolymerase Tailspike: Mechanistic Insights into Capsular Degradation. Structure 2020, 28, 613–624.e4. [Google Scholar] [CrossRef]
- Alqurashi, E.; Elbanna, K.; Ahmad, I.; Abulreesh, H.H. Antibiotic Resistance in Proteus mirabilis: Mechanism, Status, and Public Health Significance. J. Pure Appl. Microbiol. 2022, 16, 1550–1561. [Google Scholar] [CrossRef]
- Stock, I. Natural antibiotic susceptibility of Proteus spp., with special reference to P. mirabilis and P. penneri strains. J. Chemother. 2003, 15, 12–26. [Google Scholar] [CrossRef]
- Sanchez, B.C.; Heckmann, E.R.; Green, S.I.; Clark, J.R.; Kaplan, H.B.; Ramig, R.F.; Hines-Munson, C.; Skelton, F.; Trautner, B.W.; Maresso, A.W. Development of Phage Cocktails to Treat E. coli Catheter-Associated Urinary Tract Infection and Associated Biofilms. Front. Microbiol. 2022, 13, 796132. [Google Scholar] [CrossRef]
- Terwiliger, A.; Clark, J.; Karris, M.; Hernandez-Santos, H.; Green, S.; Aslam, S.; Maresso, A. Phage Therapy Related Microbial Succession Associated with Successful Clinical Outcome for a Recurrent Urinary Tract Infection. Viruses 2021, 13, 2049. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Strain | BigMira | MidiMira |
|---|---|---|
| Proteus mirabilis 074 | − | − |
| Proteus mirabilis 082 | − | − |
| Proteus mirabilis 114 | − | − |
| Proteus mirabilis 129 | − | − |
| Proteus mirabilis 159 | − | − |
| Proteus mirabilis 163 | − | − |
| Proteus mirabilis 195 | − | − |
| Proteus mirabilis 204 | − | − |
| Proteus mirabilis 218 | − | − |
| Proteus mirabilis MCS | + | + |
| Proteus mirabilis 5460 | − | − |
| Proteus Phage | Host Range (Proteus spp.) | Burst Size | Genome Length (bp) | G + C Content (%) | Putative CDSs |
|---|---|---|---|---|---|
| PM 85 [56] | 3/30 | 18 | 43,642 | 39.3 | 47 |
| PM 93 [56] | 2/30 | 75 | 45,169 | 39.4 | 48 |
| PM 116 [56] | 2/30 | 70 | 44,601 | 39.2 | 53 |
| Pm 5460 [36] | 16/26 | 46 | 44,573 | 39.6 | 56 |
| BigMira | 1/11 | 13 | 43,026 | 39.4 | 52 |
| MidiMira | 1/11 | 39 | 43,026 | 39.4 | 52 |
| BigMira-UFV01 | MidiMira-UFV02 | Nucleo-Tide Position | Altered Protein | ||||
|---|---|---|---|---|---|---|---|
| Nucleo-Tide | Amino Acid | Classification | Nucleo-Tide | Amino Acid | Classification | ||
| A | Y (Tyrosine) | Hydrophobic aromatic | G | C (Cysteine) | Hydrophilic uncharged | 40,548 | Phage tail fiber (gp51) |
| G | G (Glycine) | Hydrophobic aliphatic | A | S (Serine) | Hydrophilic uncharged | 41,135 | |
| G | D (Aspartic acid) | Hydrophilic Acidic | A | N (Aspara-gine) | Hydrophilic uncharged | 41,141 | |
| C | F (Phenyl-alanine) | Hydrophobic aromatic | A | L (Leucine) | Hydrophobic aliphatic | 41,275 | |
| Bacterial Strain | Number of AMR Genes | Gene Name | Antimicrobial Class |
|---|---|---|---|
| P. mirabilis MCS | 12 | sulI | sulfonamides |
| catA2 | chloramphenicol | ||
| vat | streptogramins | ||
| dfrA1 | diaminopyramidines | ||
| qacEdelta1 | antiseptics | ||
| tetQ tetA | tetracyclines | ||
| blaOXA-9 blaCTX-M-2 | β-lactams | ||
| aac(6′)-Iq aac(6′)-Ib’ aadA | aminoglycosides | ||
| P. mirabilis 204 | 8 | sulI | sulfonamides |
| tetA | tetracyclines | ||
| blaTEM-135 | β-lactams | ||
| aadA aadA2 aph(3′)-Ia aph(6)-Id aph(3″)-Ib | aminoglycosides | ||
| P. mirabilis 195 | 4 | catII | chloramphenicol |
| vat | streptogramins | ||
| dfrA1 | diaminopyramidines | ||
| aadA | aminoglycosides | ||
| P. mirabilis 114 P. mirabilis 159 P. mirabilis 163 | 2 | vat | streptogramins |
| dfrA1 | diaminopyramidines | ||
| P. mirabilis 074 | 1 | qnrD1 | fluoroquinolones |
| P. mirabilis 082 | 1 | tetQ | tetracyclines |
| P. mirabilis 129 | 1 | blaTEM-2 | β-lactams |
| P. mirabilis 218 | 1 | blaTEM-2 | β-lactams |
| Core genes: crp, kpnH, gyrB, rsmA, and catA4 | multidrug efflux pump, quinolones, chloramphenicol | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, J.D.; Bens, L.; Santos, A.J.d.C.; Lavigne, R.; Soares, J.; Melo, L.D.R.; Vallino, M.; Dias, R.S.; Drulis-Kawa, Z.; de Paula, S.O.; et al. Isolation and Characterization of the Acadevirus Members BigMira and MidiMira Infecting a Highly Pathogenic Proteus mirabilis Strain. Microorganisms 2023, 11, 2141. https://doi.org/10.3390/microorganisms11092141
da Silva JD, Bens L, Santos AJdC, Lavigne R, Soares J, Melo LDR, Vallino M, Dias RS, Drulis-Kawa Z, de Paula SO, et al. Isolation and Characterization of the Acadevirus Members BigMira and MidiMira Infecting a Highly Pathogenic Proteus mirabilis Strain. Microorganisms. 2023; 11(9):2141. https://doi.org/10.3390/microorganisms11092141
Chicago/Turabian Styleda Silva, Jéssica Duarte, Lene Bens, Adriele J. do Carmo Santos, Rob Lavigne, José Soares, Luís D. R. Melo, Marta Vallino, Roberto Sousa Dias, Zuzanna Drulis-Kawa, Sérgio Oliveira de Paula, and et al. 2023. "Isolation and Characterization of the Acadevirus Members BigMira and MidiMira Infecting a Highly Pathogenic Proteus mirabilis Strain" Microorganisms 11, no. 9: 2141. https://doi.org/10.3390/microorganisms11092141