
Chronic Granulomatous Disease (CGD): Commonly Associated Pathogens, Diagnosis and Treatment
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Pathogenesis
3. Clinical Manifestations
3.1. CGD-Related Inflammatory Responses
3.2. Hemophagocytic Lymphohistiocytosis (HLH)
4. CGD Incidence
5. Granulomas
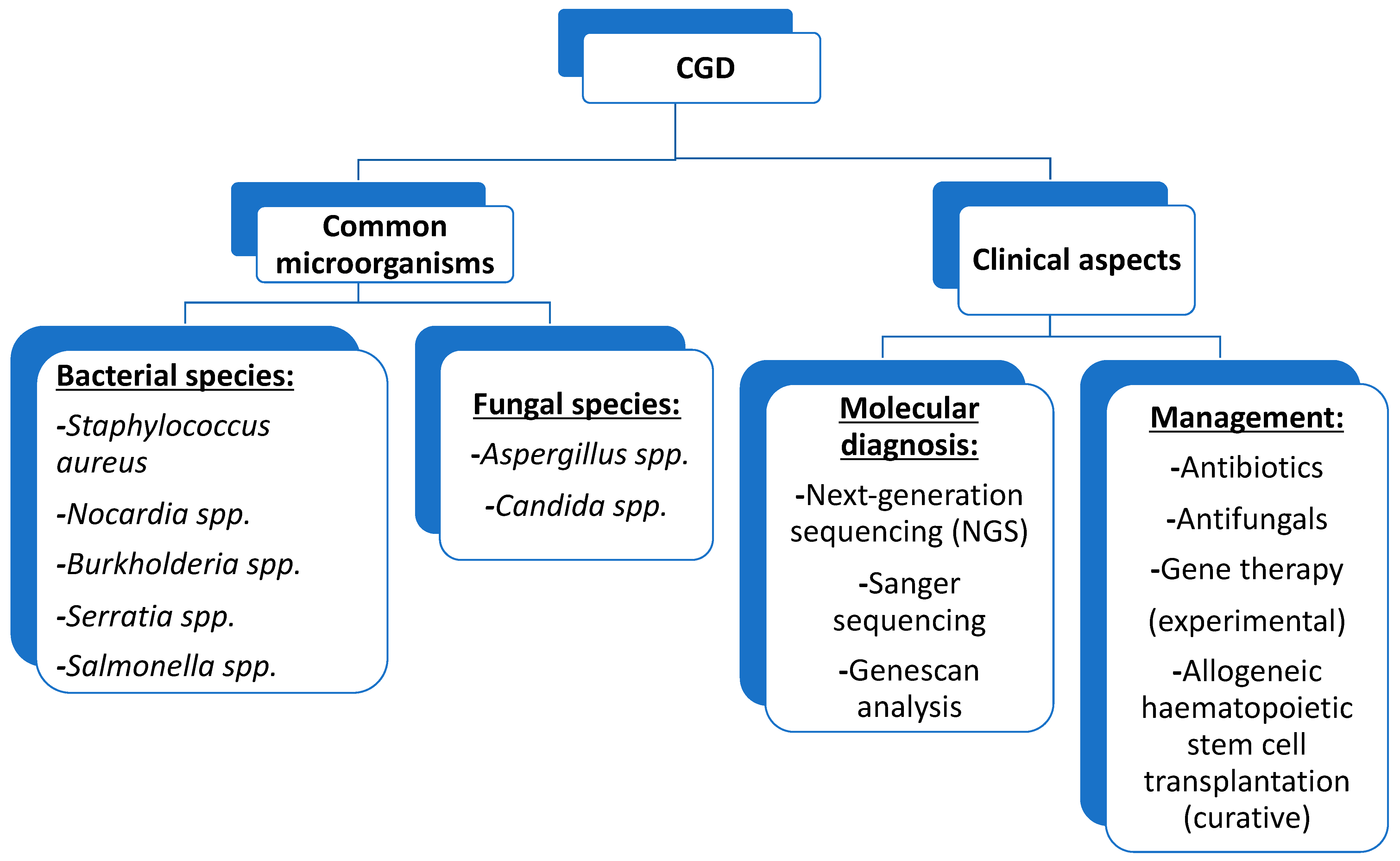
6. Pathogens and CGD-Related Infectious Diseases
6.1. Pathogens
6.2. CGD-Related Infectious Diseases
6.2.1. Lung Involvement
6.2.2. Skin Site Involvement
6.2.3. Liver Involvement
6.2.4. Gastrointestinal Tract Involvement
6.2.5. Bone Involvement
6.2.6. Other Involvements
7. Laboratory Diagnosis
7.1. Neutrophil-Function Testing
7.2. Nitroblue Tetrazolium (NBT) Reduction Test
7.3. Flow Cytometric Dihydrorhodamine Assay
7.4. Luminol-Enhanced Chemiluminescence Assay
7.5. Genetic Testing
8. Management of CGD
8.1. Haematopoietic Stem Cell Transplantation (HSCT/HCT)
8.2. Drug-Based Treatment
8.3. Gene Therapy
8.4. Other Therapies
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bridges, R.A.; Berendes, H.; Good, R.A. A Fatal Granulomatous Disease of Childhood; the Clinical, Pathological, and Laboratory Features of a New Syndrome. AMA J. Dis. Child. 1959, 97, 387–408. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, P.A. Chronic Granulomatous Disease of Childhood. Perspect. Pediatr. Pathol. 1982, 7, 237–258. [Google Scholar] [PubMed]
- Good, R.A.; Quie, P.G.; Windhorst, D.B.; Page, A.R.; Rodey, G.E.; White, J.; Wolfson, J.J.; Holmes, B.H. Fatal (chronic) Granulomatous Disease of Childhood: A Hereditary Defect of Leukocyte Function. Semin. Hematol. 1968, 5, 215–254. [Google Scholar] [PubMed]
- Anjani, G.; Vignesh, P.; Joshi, V.; Shandilya, J.K.; Bhattarai, D.; Sharma, J.; Rawat, A. Recent Advances in Chronic Granulomatous Disease. Genes Dis. 2020, 7, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Roos, D. Chronic Granulomatous Disease. Br. Med. Bull. 2016, 118, 50–63. [Google Scholar] [CrossRef]
- Mollin, M.; Beaumel, S.; Vigne, B.; Brault, J.; Roux-Buisson, N.; Rendu, J.; Barlogis, V.; Catho, G.; Dumeril, C.; Fouyssac, F.; et al. Clinical, Functional and Genetic Characterization of 16 Patients Suffering from Chronic Granulomatous Disease Variants—Identification of 11 Novel Mutations in CYBB. Clin. Exp. Immunol. 2021, 203, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C. Inflammatory Consequences of Inherited Disorders Affecting Neutrophil Function. Blood 2019, 133, 2130–2139. [Google Scholar] [CrossRef]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.R.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between Host Defence, Immune Modulation, and Tissue Injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef]
- Rider, N.L.; Jameson, M.B.; Creech, C.B. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and Genetic Basis of Disease. J. Pediatr. Infect. Dis. Soc. 2018, 7, S2–S5. [Google Scholar] [CrossRef]
- Mauch, L.; Lun, A.; O’Gorman, M.R.G.; Harris, J.S.; Schulze, I.; Zychlinsky, A.; Fuchs, T.; Oelschlägel, U.; Brenner, S.; Kutter, D.; et al. Chronic Granulomatous Disease (CGD) and Complete Myeloperoxidase Deficiency Both Yield Strongly Reduced Dihydrorhodamine 123 Test Signals but Can Be Easily Discerned in Routine Testing for CGD. Clin. Chem. 2007, 53, 890–896. [Google Scholar] [CrossRef]
- Curnutte, J.T.; Scott, P.J.; Mayo, L.A. Cytosolic Components of the Respiratory Burst Oxidase: Resolution of Four Components, Two of Which Are Missing in Complementing Types of Chronic Granulomatous Disease. Proc. Natl. Acad. Sci. USA 1989, 86, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.C.; Charbonnier, L.-M.; Schejtman, A.; Aldhekri, H.; Coomber, E.L.; Dufficy, E.R.; Beenken, A.E.; Lee, J.C.; Clare, S.; Speak, A.O.; et al. EROS/CYBC1 Mutations: Decreased NADPH Oxidase Function and Chronic Granulomatous Disease. J. Allergy Clin. Immunol. 2019, 143, 782–785.e1. [Google Scholar] [CrossRef] [PubMed]
- Roos, D. Chronic Granulomatous Disease. In NADPH Oxidases: Methods and Protocols; Knaus, U.G., Leto, T.L., Eds.; Springer: New York, NY, USA, 2019; pp. 531–542. ISBN 9781493994243. [Google Scholar]
- Yu, H.-H.; Yang, Y.-H.; Chiang, B.-L. Chronic Granulomatous Disease: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2021, 61, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Valentine, G.; Thomas, T.A.; Nguyen, T.; Lai, Y.-C. Chronic Granulomatous Disease Presenting as Hemophagocytic Lymphohistiocytosis: A Case Report. Pediatrics 2014, 134, e1727–e1730. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common Severe Infections in Chronic Granulomatous Disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef]
- Wolach, B.; Gavrieli, R.; de Boer, M.; Gottesman, G.; Ben-Ari, J.; Rottem, M.; Schlesinger, Y.; Grisaru-Soen, G.; Etzioni, A.; Roos, D. Chronic Granulomatous Disease in Israel: Clinical, Functional and Molecular Studies of 38 Patients. Clin. Immunol. 2008, 129, 103–114. [Google Scholar] [CrossRef]
- Fattahi, F.; Badalzadeh, M.; Sedighipour, L.; Movahedi, M.; Fazlollahi, M.R.; Mansouri, S.D.; Khotaei, G.T.; Bemanian, M.H.; Behmanesh, F.; Hamidieh, A.A.; et al. Inheritance Pattern and Clinical Aspects of 93 Iranian Patients with Chronic Granulomatous Disease. J. Clin. Immunol. 2011, 31, 792–801. [Google Scholar] [CrossRef]
- Kutukculer, N.; Aykut, A.; Karaca, N.E.; Durmaz, A.; Aksu, G.; Genel, F.; Pariltay, E.; Cogulu, Ö.; Azarsız, E. Chronic Granulamatous Disease: Two Decades of Experience from a Paediatric Immunology Unit in a Country with High Rate of Consangineous Marriages. Scand. J. Immunol. 2019, 89, e12737. [Google Scholar] [CrossRef]
- Wolach, B.; Gavrieli, R.; de Boer, M.; van Leeuwen, K.; Berger-Achituv, S.; Stauber, T.; Ben Ari, J.; Rottem, M.; Schlesinger, Y.; Grisaru-Soen, G.; et al. Chronic Granulomatous Disease: Clinical, Functional, Molecular, and Genetic Studies. The Israeli Experience with 84 Patients. Am. J. Hematol. 2017, 92, 28–36. [Google Scholar] [CrossRef]
- van den Berg, J.M.; van Koppen, E.; Ahlin, A.; Belohradsky, B.H.; Bernatowska, E.; Corbeel, L.; Español, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic Granulomatous Disease: The European Experience. PLoS ONE 2009, 4, e5234. [Google Scholar] [CrossRef]
- Barkai, T.; Somech, R.; Broides, A.; Gavrieli, R.; Wolach, B.; Marcus, N.; Hagin, D.; Stauber, T. Late Diagnosis of Chronic Granulomatous Disease. Clin. Exp. Immunol. 2020, 201, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic Granulomatous Disease. Report on a National Registry of 368 Patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Slack, M.A.; Thomsen, I.P. Prevention of Infectious Complications in Patients With Chronic Granulomatous Disease. J. Pediatr. Infect. Dis. Soc. 2018, 7, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Alimchandani, M.; Lai, J.-P.; Aung, P.P.; Khangura, S.; Kamal, N.; Gallin, J.I.; Holland, S.M.; Malech, H.L.; Heller, T.; Miettinen, M.; et al. Gastrointestinal Histopathology in Chronic Granulomatous Disease: A Study of 87 Patients. Am. J. Surg. Pathol. 2013, 37, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Dotis, J.; Pana, Z.D.; Roilides, E. Non-Aspergillus Fungal Infections in Chronic Granulomatous Disease. Mycoses 2013, 56, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Haidar, G.; Zerbe, C.S.; Cheng, M.; Zelazny, A.M.; Holland, S.M.; Sheridan, K.R. Phellinus Species: An Emerging Cause of Refractory Fungal Infections in Patients with X-Linked Chronic Granulomatous Disease. Mycoses 2017, 60, 155–160. [Google Scholar] [CrossRef]
- Angelino, G.; De Angelis, P.; Faraci, S.; Rea, F.; Romeo, E.F.; Torroni, F.; Tambucci, R.; Claps, A.; Francalanci, P.; Chiriaco, M.; et al. Inflammatory Bowel Disease in Chronic Granulomatous Disease: An Emerging Problem over a Twenty Years’ Experience. Pediatr. Allergy Immunol. 2017, 28, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Kadaria, D.; Sodhi, A.; Fox, R.; Williams, G.; Threlkeld, S. Chronic Granulomatous Disease Presenting as Aspergillus fumigatus Pneumonia in a Previously Healthy Young Woman. Am. J. Case Rep. 2017, 18, 351–354. [Google Scholar] [CrossRef]
- Shigemura, T.; Nakazawa, Y.; Amano, Y.; Sudo, A.; Watanabe, M.; Kobayashi, M.; Kobayashi, N.; Koike, K.; Agematsu, K.; Nishimura, K. Subcutaneous Abscess due to the Basidiomycete Phellinus mori in a Patient with Chronic Granulomatous Disease. Infection 2015, 43, 371–375. [Google Scholar] [CrossRef]
- Kobayashi, S.; Murayama, S.; Takanashi, S.; Takahashi, K.; Miyatsuka, S.; Fujita, T.; Ichinohe, S.; Koike, Y.; Kohagizawa, T.; Mori, H.; et al. Clinical Features and Prognoses of 23 Patients with Chronic Granulomatous Disease Followed for 21 Years by a Single Hospital in Japan. Eur. J. Pediatr. 2008, 167, 1389–1394. [Google Scholar] [CrossRef]
- Mortaz, E.; Sarhifynia, S.; Marjani, M.; Moniri, A.; Mansouri, D.; Mehrian, P.; van Leeuwen, K.; Roos, D.; Garssen, J.; Adcock, I.M.; et al. An Adult Autosomal Recessive Chronic Granulomatous Disease Patient with Pulmonary Aspergillus terreus Infection. BMC Infect. Dis. 2018, 18, 552. [Google Scholar] [CrossRef]
- Kaltenis, P.; Mudeniené, V.; Maknavicius, S.; Seinin, D. Renal Amyloidosis in a Child with Chronic Granulomatous Disease and Invasive Aspergillosis. Pediatr. Nephrol. 2008, 23, 831–834. [Google Scholar] [CrossRef]
- Siddiqui, S.; Anderson, V.L.; Hilligoss, D.M.; Abinun, M.; Kuijpers, T.W.; Masur, H.; Witebsky, F.G.; Shea, Y.R.; Gallin, J.I.; Malech, H.L.; et al. Fulminant Mulch Pneumonitis: An Emergency Presentation of Chronic Granulomatous Disease. Clin. Infect. Dis. 2007, 45, 673–681. [Google Scholar] [CrossRef]
- Battersby, A.C.; Cale, A.M.; Goldblatt, D.; Gennery, A.R. Clinical Manifestations of Disease in X-Linked Carriers of Chronic Granulomatous Disease. J. Clin. Immunol. 2013, 33, 1276–1284. [Google Scholar] [CrossRef]
- Thomsen, I.P.; Smith, M.A.; Holland, S.M.; Creech, C.B. A Comprehensive Approach to the Management of Children and Adults with Chronic Granulomatous Disease. J. Allergy Clin. Immunol. Pract. 2016, 4, 1082–1088. [Google Scholar] [CrossRef]
- Köker, M.Y.; Camcıoğlu, Y.; van Leeuwen, K.; Kılıç, S.Ş.; Barlan, I.; Yılmaz, M.; Metin, A.; de Boer, M.; Avcılar, H.; Patıroğlu, T.; et al. Clinical, Functional, and Genetic Characterization of Chronic Granulomatous Disease in 89 Turkish Patients. J. Allergy Clin. Immunol. 2013, 132, 1156–1163.e5. [Google Scholar] [CrossRef]
- Dotis, J.; Roilides, E. Osteomyelitis due to Aspergillus spp. in Patients with Chronic Granulomatous Disease: Comparison of Aspergillus nidulans and Aspergillus fumigatus. Int. J. Infect. Dis. 2004, 8, 103–110. [Google Scholar] [CrossRef]
- Marciano, B.E.; Rosenzweig, S.D.; Kleiner, D.E.; Anderson, V.L.; Darnell, D.N.; Anaya-O’Brien, S.; Hilligoss, D.M.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Gastrointestinal Involvement in Chronic Granulomatous Disease. Pediatrics 2004, 114, 462–468. [Google Scholar] [CrossRef]
- Norouzi, S.; Aghamohammadi, A.; Mamishi, S.; Rosenzweig, S.D.; Rezaei, N. Bacillus Calmette-Guérin (BCG) Complications Associated with Primary Immunodeficiency Diseases. J. Infect. 2012, 64, 543–554. [Google Scholar] [CrossRef]
- Shin, K.-S.; Lee, M.S. Concomitant Use of Corticosteroid and Antimicrobials for Liver Abscesses in Patients with Chronic Granulomatous Disease. Korean J. Pediatr. 2016, 59, 196–201. [Google Scholar] [CrossRef]
- Lublin, M.; Bartlett, D.L.; Danforth, D.N.; Kauffman, H.; Gallin, J.I.; Malech, H.L.; Shawker, T.; Choyke, P.; Kleiner, D.E.; Schwartzentruber, D.J.; et al. Hepatic Abscess in Patients with Chronic Granulomatous Disease. Ann. Surg. 2002, 235, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Hussain, N.; Wright, E.C.; Kleiner, D.E.; Hoofnagle, J.H.; Ahlawat, S.; Anderson, V.; Hilligoss, D.; Gallin, J.I.; Liang, T.J.; et al. Hepatic Involvement and Portal Hypertension Predict Mortality in Chronic Granulomatous Disease. Gastroenterology 2008, 134, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Campos, A.; Scheffler-Mendoza, S.; León-Lara, X.; Onuma-Zamayoa, H.; Espinosa, S.; Yamazaki-Nakashimada, M.A.; Blancas-Galicia, L. Infectious and inflammatory gastrointestinal manifestations of chronic granulomatous disease. Rev. Alerg. Mex. 2021, 68, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Vignesh, P.; Sharma, R.; Barman, P.; Mondal, S.; Das, J.; Siniah, S.; Goyal, T.; Sharma, S.; Pilania, R.K.; Jindal, A.K.; et al. Impact of COVID-19 Pandemic on Clinical Care of Patients and Psychosocial Health of Affected Families with Chronic Granulomatous Disease: An Observational Study from North India. J. Clin. Immunol. 2023, 1–13. [Google Scholar] [CrossRef]
- Esmaeilzadeh, H.; Dehghani, S.S.; Shahhoseini, B.; Alyasin, S.; Nabavizadeh, S.H.; Askari, A. COVID-19 in Chronic Granulomatosis Disease: A Case Report. Iran. J. Allergy Asthma Immunol. 2022, 21, 478–483. [Google Scholar] [CrossRef]
- Rawat, A.; Vignesh, P.; Sudhakar, M.; Sharma, M.; Suri, D.; Jindal, A.; Gupta, A.; Shandilya, J.K.; Loganathan, S.K.; Kaur, G.; et al. Clinical, Immunological, and Molecular Profile of Chronic Granulomatous Disease: A Multi-Centric Study of 236 Patients From India. Front. Immunol. 2021, 12, 625320. [Google Scholar] [CrossRef]
- Roos, D.; de Boer, M. Molecular Diagnosis of Chronic Granulomatous Disease. Clin. Exp. Immunol. 2014, 175, 139–149. [Google Scholar] [CrossRef]
- Chen, Y.; Junger, W.G. Measurement of Oxidative Burst in Neutrophils. Methods Mol. Biol. 2012, 844, 115–124. [Google Scholar]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Verbsky, J.W.; Routes, J.M. Recurrent Fever, Immune Deficiency, and Autoinflammatory Disorders. In Nelson Pediatric Symptom-Based Diagnosis: Common Diseases and Their Mimics; Elsevier: Amsterdam, The Netherlands, 2023; pp. 1015–1046.e1. ISBN 9780323761741. [Google Scholar]
- Jirapongsananuruk, O.; Malech, H.L.; Kuhns, D.B.; Niemela, J.E.; Brown, M.R.; Anderson-Cohen, M.; Fleisher, T.A. Diagnostic Paradigm for Evaluation of Male Patients with Chronic Granulomatous Disease, Based on the Dihydrorhodamine 123 Assay. J. Allergy Clin. Immunol. 2003, 111, 374–379. [Google Scholar] [CrossRef]
- Kuhns, D.B.; Alvord, W.G.; Heller, T.; Feld, J.J.; Pike, K.M.; Marciano, B.E.; Uzel, G.; DeRavin, S.S.; Priel, D.A.L.; Soule, B.P.; et al. Residual NADPH Oxidase and Survival in Chronic Granulomatous Disease. N. Engl. J. Med. 2010, 363, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Galicia, L.; Santos-Chávez, E.; Deswarte, C.; Mignac, Q.; Medina-Vera, I.; León-Lara, X.; Roynard, M.; Scheffler-Mendoza, S.C.; Rioja-Valencia, R.; Alvirde-Ayala, A.; et al. Genetic, Immunological, and Clinical Features of the First Mexican Cohort of Patients with Chronic Granulomatous Disease. J. Clin. Immunol. 2020, 40, 475–493. [Google Scholar] [CrossRef]
- Sanabria, D.; Giménez, V.; de Cuéllar, C.M.; Carpinelli, M.; Benegas, S.; Insaurralde, S. Enfermedad Granulomatosa Crónica. Diagnóstico Mediante El Ensayo de Dihidrorodamina. Rev. Chil. Pediatría 2020, 91, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Ang, E.Y.; Soh, J.Y.; Liew, W.K.; Chan, K.W.; Thoon, K.C.; Chong, C.Y.; Lau, Y.L.; Lee, B.W. Reliability of Acute Illness Dihydrorhodamine-123 Testing for Chronic Granulomatous Disease. Clin. Lab. 2013, 59, 203–206. [Google Scholar]
- Lai, B.; Bernhardt, P.V.; Krömer, J.O. Cytochrome c Reductase Is a Key Enzyme Involved in the Extracellular Electron Transfer Pathway towards Transition Metal Complexes in Pseudomonas putida. ChemSusChem 2020, 13, 5308–5317. [Google Scholar] [CrossRef]
- Yu, J.E.; Azar, A.E.; Chong, H.J.; Jongco, A.M., 3rd; Prince, B.T. Considerations in the Diagnosis of Chronic Granulomatous Disease. J. Pediatr. Infect. Dis. Soc. 2018, 7, S6–S11. [Google Scholar] [CrossRef]
- Vowells, S.J.; Sekhsaria, S.; Malech, H.L.; Shalit, M.; Fleisher, T.A. Flow Cytometric Analysis of the Granulocyte Respiratory Burst: A Comparison Study of Fluorescent Probes. J. Immunol. Methods 1995, 178, 89–97. [Google Scholar] [CrossRef]
- Leiding, J.W.; Holland, S.M. Chronic Granulomatous Disease; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Parvaneh, N.; Teimourian, S. Effectiveness of Nitroblue Tetrazolium (NBT) Test. Arch. Iran. Med. 2008, 11, 129–130; author reply 130. [Google Scholar] [PubMed]
- Molehin, A.J.; Nichols, J.; Smith, F.; Nugent, K. Phagocytosis: Biology and Methods. In Encyclopedia of Infection and Immunity; Rezaei, N., Ed.; Elsevier: Oxford, UK, 2022; pp. 134–140. ISBN 9780323903035. [Google Scholar]
- Güngör, T.; Teira, P.; Slatter, M.; Stussi, G.; Stepensky, P.; Moshous, D.; Vermont, C.; Ahmad, I.; Shaw, P.J.; Telles da Cunha, J.M.; et al. Reduced-Intensity Conditioning and HLA-Matched Haemopoietic Stem-Cell Transplantation in Patients with Chronic Granulomatous Disease: A Prospective Multicentre Study. Lancet 2014, 383, 436–448. [Google Scholar] [CrossRef]
- Vowells, S.J.; Fleisher, T.A.; Sekhsaria, S.; Alling, D.W.; Maguire, T.E.; Malech, H.L. Genotype-Dependent Variability in Flow Cytometric Evaluation of Reduced Nicotinamide Adenine Dinucleotide Phosphate Oxidase Function in Patients with Chronic Granulomatous Disease. J. Pediatr. 1996, 128, 104–107. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Kim, H.-J.; Ki, C.-S.; Kim, D.W.; Yoo, K.H.; Kang, E.-S. Rapid Determination of Chimerism Status Using Dihydrorhodamine Assay in a Patient with X-Linked Chronic Granulomatous Disease Following Hematopoietic Stem Cell Transplantation. Ann. Lab. Med. 2013, 33, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C. Diagnostic Chimerism Analysis after Allogeneic Stem Cell Transplantation: New Methods and Markers. Am. J. Pharmacogenom. 2004, 4, 177–187. [Google Scholar] [CrossRef]
- Neehus, A.-L.; Tuano, K.; Le Voyer, T.; Nandiwada, S.L.; Murthy, K.; Puel, A.; Casanova, J.-L.; Chinen, J.; Bustamante, J. Chronic Granulomatous Disease-Like Presentation of a Child with Autosomal Recessive PKCδ Deficiency. J. Clin. Immunol. 2022, 42, 1244–1253. [Google Scholar] [CrossRef]
- Ríos, N.; Prolo, C.; Álvarez, M.N.; Piacenza, L.; Radi, R. Chapter 21—Peroxynitrite Formation and Detection in Living Cells. In Nitric Oxide, 3rd ed.; Ignarro, L.J., Freeman, B.A., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 271–288. ISBN 9780128042731. [Google Scholar]
- Görlach, A.; Lee, P.L.; Roesler, J.; Hopkins, P.J.; Christensen, B.; Green, E.D.; Chanock, S.J.; Curnutte, J.T. A p47-Phox Pseudogene Carries the Most Common Mutation Causing p47-Phox- Deficient Chronic Granulomatous Disease. J. Clin. Investig. 1997, 100, 1907–1918. [Google Scholar] [CrossRef]
- Kulkarni, M.; Hule, G.; de Boer, M.; van Leeuwen, K.; Kambli, P.; Aluri, J.; Gupta, M.; Dalvi, A.; Mhatre, S.; Taur, P.; et al. Approach to Molecular Diagnosis of Chronic Granulomatous Disease (CGD): An Experience from a Large Cohort of 90 Indian Patients. J. Clin. Immunol. 2018, 38, 898–916. [Google Scholar] [CrossRef]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Roesler, J.; Lopez, J.A.; Ariga, T.; Avcin, T.; de Boer, M.; Bustamante, J.; Condino-Neto, A.; et al. Hematologically Important Mutations: X-Linked Chronic Granulomatous Disease (third Update). Blood Cells Mol. Dis. 2010, 45, 246–265. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, D.B. Diagnostic Testing for Chronic Granulomatous Disease. In NADPH Oxidases: Methods and Protocols; Knaus, U.G., Leto, T.L., Eds.; Springer: New York, NY, USA, 2019; pp. 543–571. ISBN 9781493994243. [Google Scholar]
- Yonkof, J.R.; Gupta, A.; Fu, P.; Garabedian, E.; Dalal, J. The United States Immunodeficiency Network Consortium Role of Allogeneic Hematopoietic Stem Cell Transplant for Chronic Granulomatous Disease (CGD): A Report of the United States Immunodeficiency Network. J. Clin. Immunol. 2019, 39, 448–458. [Google Scholar] [CrossRef]
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef]
- Chiesa, R.; Wang, J.; Blok, H.-J.; Hazelaar, S.; Neven, B.; Moshous, D.; Schulz, A.; Hoenig, M.; Hauck, F.; Al Seraihy, A.; et al. Hematopoietic Cell Transplantation in Chronic Granulomatous Disease: A Study of 712 Children and Adults. Blood 2020, 136, 1201–1211. [Google Scholar] [CrossRef]
- Connelly, J.A.; Marsh, R.; Parikh, S.; Talano, J.-A. Allogeneic Hematopoietic Cell Transplantation for Chronic Granulomatous Disease: Controversies and State of the Art. J. Pediatr. Infect. Dis. Soc. 2018, 7, S31–S39. [Google Scholar] [CrossRef] [PubMed]
- Magnani, A.; Mahlaoui, N. Managing Inflammatory Manifestations in Patients with Chronic Granulomatous Disease. Paediatr. Drugs 2016, 18, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Yanagimachi, M.; Kato, K.; Iguchi, A.; Sasaki, K.; Kiyotani, C.; Koh, K.; Koike, T.; Sano, H.; Shigemura, T.; Muramatsu, H.; et al. Hematopoietic Cell Transplantation for Chronic Granulomatous Disease in Japan. Front. Immunol. 2020, 11, 1617. [Google Scholar] [CrossRef]
- Marsh, R.A.; Leiding, J.W.; Logan, B.R.; Griffith, L.M.; Arnold, D.E.; Haddad, E.; Falcone, E.L.; Yin, Z.; Patel, K.; Arbuckle, E.; et al. Chronic Granulomatous Disease-Associated IBD Resolves and Does Not Adversely Impact Survival Following Allogeneic HCT. J. Clin. Immunol. 2019, 39, 653–667. [Google Scholar] [CrossRef]
- Yi, E.S.; Choi, Y.B.; Lee, N.H.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Kang, E.-S.; Kim, Y.-J.; Yoo, K.H. Allogeneic Hematopoietic Cell Transplantation in Patients with Primary Immunodeficiencies in Korea: Eleven-Year Experience in a Single Center. J. Clin. Immunol. 2018, 38, 757–766. [Google Scholar] [CrossRef]
- Mosaad, Y.M. Hematopoietic Stem Cells: An Overview. Transfus. Apher. Sci. 2014, 51, 68–82. [Google Scholar] [CrossRef]
- Hawley, R.G.; Ramezani, A.; Hawley, T.S. Hematopoietic Stem Cells. Methods Enzymol. 2006, 419, 149–179. [Google Scholar] [PubMed]
- Garcia-Eulate, R.; Hussain, N.; Heller, T.; Kleiner, D.; Malech, H.; Holland, S.; Choyke, P.L. CT and MRI of Hepatic Abscess in Patients with Chronic Granulomatous Disease. AJR Am. J. Roentgenol. 2006, 187, 482–490. [Google Scholar] [CrossRef]
- Kohn, D.B.; Booth, C.; Kang, E.M.; Pai, S.-Y.; Shaw, K.L.; Santilli, G.; Armant, M.; Buckland, K.F.; Choi, U.; De Ravin, S.S.; et al. Lentiviral Gene Therapy for X-Linked Chronic Granulomatous Disease. Nat. Med. 2020, 26, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Pontes, R.; Gomes, L.N.; de Albuquerque, R.S.; Soeiro-Pereira, P.V.; Condino-Neto, A. The Extended Understanding of Chronic Granulomatous Disease. Curr. Opin. Pediatr. 2019, 31, 869–873. [Google Scholar] [CrossRef]
- Chiriaco, M.; Salfa, I.; Di Matteo, G.; Rossi, P.; Finocchi, A. Chronic Granulomatous Disease: Clinical, Molecular, and Therapeutic Aspects. Pediatr. Allergy Immunol. 2016, 27, 242–253. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kühlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-Linked Chronic Granulomatous Disease by Gene Therapy, Augmented by Insertional Activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Kang, E.M.; Choi, U.; Theobald, N.; Linton, G.; Long Priel, D.A.; Kuhns, D.; Malech, H.L. Retrovirus Gene Therapy for X-Linked Chronic Granulomatous Disease Can Achieve Stable Long-Term Correction of Oxidase Activity in Peripheral Blood Neutrophils. Blood 2010, 115, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Edvard Smith, C.I.; Puck, J. Primary Immunodeficiency Diseases: A Molecular and Genetic Approach; Oxford University Press: Oxford, UK, 2007; ISBN 9780195147742. [Google Scholar]
- Conrad, A.; Neven, B.; Mahlaoui, N.; Suarez, F.; Sokol, H.; Ruemmele, F.M.; Rouzaud, C.; Moshous, D.; Lortholary, O.; Blanche, S.; et al. Infections in Patients with Chronic Granulomatous Disease Treated with Tumor Necrosis Factor Alpha Blockers for Inflammatory Complications. J. Clin. Immunol. 2021, 41, 185–193. [Google Scholar] [CrossRef]
- Peixoto, A.; Coelho, R.; Maia, T.; Sarmento, A.; Magro, F.; Macedo, G. Chronic Granulomatous Disease Mimicking Colonic Crohn’s Disease Successfully Treated with Infliximab. ACG Case Rep. J. 2017, 4, e46. [Google Scholar] [CrossRef] [PubMed]
- Straughan, D.M.; McLoughlin, K.C.; Mullinax, J.E.; Marciano, B.E.; Freeman, A.F.; Anderson, V.L.; Uzel, G.; Azoury, S.C.; Sorber, R.; Quadri, H.S.; et al. The Changing Paradigm of Management of Liver Abscesses in Chronic Granulomatous Disease. Clin. Infect. Dis. 2018, 66, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Al Ghadeer, H.A.; Busaleh, F.N.; Al Habeeb, J.A.; Alaithan, R.M.; Almutahhar, A.E.; Bin Abd, M.M.; Aldawood, M.M. Liver Abscesses as a Sign of Chronic Granulomatous Disease in Adolescent. Cureus 2021, 13, e17467. [Google Scholar] [CrossRef]
- Mortaz, E.; Azempour, E.; Mansouri, D.; Tabarsi, P.; Ghazi, M.; Koenderman, L.; Roos, D.; Adcock, I.M. Common Infections and Target Organs Associated with Chronic Granulomatous Disease in Iran. Int. Arch. Allergy Immunol. 2019, 179, 62–73. [Google Scholar] [CrossRef]
- Pilania, R.K.; Rawat, A.; Vignesh, P.; Guleria, S.; Jindal, A.K.; Das, G.; Suri, D.; Gupta, A.; Gupta, K.; Chan, K.-W.; et al. Liver Abscess in Chronic Granulomatous Disease-Two Decades of Experience from a Tertiary Care Centre in North-West India. J. Clin. Immunol. 2021, 41, 552–564. [Google Scholar] [CrossRef]
- Kang, E.M.; Malech, H.L. Gene Therapy for Chronic Granulomatous Disease. Methods Enzymol. 2012, 507, 125–154. [Google Scholar]
- Santilli, G.; Almarza, E.; Brendel, C.; Choi, U.; Beilin, C.; Blundell, M.P.; Haria, S.; Parsley, K.L.; Kinnon, C.; Malech, H.L.; et al. Biochemical Correction of X-CGD by a Novel Chimeric Promoter Regulating High Levels of Transgene Expression in Myeloid Cells. Mol. Ther. 2011, 19, 122–132. [Google Scholar] [CrossRef]
- Kang, H.J.; Bartholomae, C.C.; Paruzynski, A.; Arens, A.; Kim, S.; Yu, S.S.; Hong, Y.; Joo, C.-W.; Yoon, N.-K.; Rhim, J.-W.; et al. Retroviral Gene Therapy for X-Linked Chronic Granulomatous Disease: Results from Phase I/II Trial. Mol. Ther. 2011, 19, 2092–2101. [Google Scholar] [CrossRef]
- Dedieu, C.; Albert, M.H.; Mahlaoui, N.; Hauck, F.; Hedrich, C.; Baumann, U.; Warnatz, K.; Roesler, J.; Speckmann, C.; Schulte, J.; et al. Outcome of Chronic Granulomatous Disease—Conventional Treatment vs Stem Cell Transplantation. Pediatr. Allergy Immunol. 2021, 32, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Åhlin, A.; Fugeläng, J.; de Boer, M.; Ringden, O.; Fasth, A.; Winiarski, J. Chronic Granulomatous Disease-Haematopoietic Stem Cell Transplantation versus Conventional Treatment. Acta Paediatr. 2013, 102, 1087–1094. [Google Scholar] [CrossRef]
- Baha, A.; Hanazay, C.; Kokturk, N.; Turktas, H. A Case of Sarcoidosis Associated With Anti-Tumor Necrosis Factor Treatment. J. Investig. Med. High Impact Case Rep. 2015, 3, 2324709615571366. [Google Scholar] [CrossRef]
- Yang, A.H.; Sullivan, B.; Zerbe, C.S.; De Ravin, S.S.; Blakely, A.M.; Quezado, M.M.; Marciano, B.E.; Marko, J.; Ling, A.; Kleiner, D.E.; et al. Gastrointestinal and Hepatic Manifestations of Chronic Granulomatous Disease. J. Allergy Clin. Immunol. Pract. 2023, 11, 1401–1416. [Google Scholar] [CrossRef] [PubMed]
- Lugo Reyes, S.O.; González Garay, A.; González Bobadilla, N.Y.; Rivera Lizárraga, D.A.; Madrigal Paz, A.C.; Medina-Torres, E.A.; Álvarez Cardona, A.; Galindo Ortega, J.L.; Solís Galicia, C.; Espinosa-Padilla, S.E.; et al. Efficacy and Safety of Interferon-Gamma in Chronic Granulomatous Disease: A Systematic Review and Meta-Analysis. J. Clin. Immunol. 2023, 43, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Rothe, M.; Santilli, G.; Charrier, S.; Stein, S.; Kunkel, H.; Abriss, D.; Müller-Kuller, U.; Gaspar, B.; Modlich, U.; et al. Non-Clinical Efficacy and Safety Studies on G1XCGD, a Lentiviral Vector for Ex Vivo Gene Therapy of X-Linked Chronic Granulomatous Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 69–79. [Google Scholar] [CrossRef]
- Jofra Hernández, R.; Calabria, A.; Sanvito, F.; De Mattia, F.; Farinelli, G.; Scala, S.; Visigalli, I.; Carriglio, N.; De Simone, M.; Vezzoli, M.; et al. Hematopoietic Tumors in a Mouse Model of X-Linked Chronic Granulomatous Disease after Lentiviral Vector-Mediated Gene Therapy. Mol. Ther. 2021, 29, 86–102. [Google Scholar] [CrossRef]
- Farinelli, G.; Jofra Hernandez, R.; Rossi, A.; Ranucci, S.; Sanvito, F.; Migliavacca, M.; Brombin, C.; Pramov, A.; Di Serio, C.; Bovolenta, C.; et al. Lentiviral Vector Gene Therapy Protects XCGD Mice From Acute Staphylococcus Aureus Pneumonia and Inflammatory Response. Mol. Ther. 2016, 24, 1873–1880. [Google Scholar] [CrossRef]
- Jafarian, A.; Shokri, G.; Shokrollahi Barough, M.; Moin, M.; Pourpak, Z.; Soleimani, M. Recent Advances in Gene Therapy and Modeling of Chronic Granulomatous Disease. Iran. J. Allergy Asthma Immunol. 2019, 18, 131–142. [Google Scholar] [CrossRef]
- Renga, G.; Oikonomou, V.; Moretti, S.; Stincardini, C.; Bellet, M.M.; Pariano, M.; Bartoli, A.; Brancorsini, S.; Mosci, P.; Finocchi, A.; et al. Thymosin β4 Promotes Autophagy and Repair via HIF-1α Stabilization in Chronic Granulomatous Disease. Life Sci. Alliance 2019, 2, e201900432. [Google Scholar] [CrossRef] [PubMed]
- Kanariou, M.; Spanou, K.; Tantou, S. Long-Term Observational Studies of Chronic Granulomatous Disease. Curr. Opin. Hematol. 2018, 25, 7–12. [Google Scholar] [CrossRef]
- Flynn, R.; Grundmann, A.; Renz, P.; Hänseler, W.; James, W.S.; Cowley, S.A.; Moore, M.D. CRISPR-Mediated Genotypic and Phenotypic Correction of a Chronic Granulomatous Disease Mutation in Human iPS Cells. Exp. Hematol. 2015, 43, 838–848.e3. [Google Scholar] [CrossRef] [PubMed]
- Malech, H.L.; Maples, P.B.; Whiting-Theobald, N.; Linton, G.F.; Sekhsaria, S.; Vowells, S.J.; Li, F.; Miller, J.A.; DeCarlo, E.; Holland, S.M.; et al. Prolonged Production of NADPH Oxidase-Corrected Granulocytes after Gene Therapy of Chronic Granulomatous Disease. Proc. Natl. Acad. Sci. USA 1997, 94, 12133–12138. [Google Scholar] [CrossRef]
- Mukherjee, S.; Thrasher, A.J. Gene Therapy for PIDs: Progress, Pitfalls and Prospects. Gene 2013, 525, 174–181. [Google Scholar] [CrossRef]
- Renga, G.; Oikonomou, V.; Stincardini, C.; Pariano, M.; Borghi, M.; Costantini, C.; Bartoli, A.; Garaci, E.; Goldstein, A.L.; Romani, L. Thymosin β4 Limits Inflammation through Autophagy. Expert Opin. Biol. Ther. 2018, 18, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Gennery, A.R. Progress in Treating Chronic Granulomatous Disease. Br. J. Haematol. 2021, 192, 251–264. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Site of Disease | Number of Episodes | Number of Patients with ≥1 Episode | % of Patients with ≥1 Episode |
|---|---|---|---|
| Lung | 634 | 284 | 66% |
| Skin/Subcutis | 341 | 299 | 53% |
| Lymph node | 622 | 213 | 50% |
| Gastro-intestinal | 643 | 208 | 48% |
| Liver | 240 | 138 | 32% |
| Kidney/ Urinary tract | 139 | 95 | 22% |
| Septicaemia | 111 | 85 | 20% |
| Ear | 84 | 62 | 14% |
| Bone | 84 | 56 | 13% |
| Eye | 68 | 46 | 11% |
| Joint | 35 | 31 | 7% |
| Brain | 34 | 31 | 7% |
| Autoimmunity-Rheumatology | 26 | 26 | 6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Justiz-Vaillant, A.A.; Williams-Persad, A.F.-A.; Arozarena-Fundora, R.; Gopaul, D.; Soodeen, S.; Asin-Milan, O.; Thompson, R.; Unakal, C.; Akpaka, P.E. Chronic Granulomatous Disease (CGD): Commonly Associated Pathogens, Diagnosis and Treatment. Microorganisms 2023, 11, 2233. https://doi.org/10.3390/microorganisms11092233
Justiz-Vaillant AA, Williams-Persad AF-A, Arozarena-Fundora R, Gopaul D, Soodeen S, Asin-Milan O, Thompson R, Unakal C, Akpaka PE. Chronic Granulomatous Disease (CGD): Commonly Associated Pathogens, Diagnosis and Treatment. Microorganisms. 2023; 11(9):2233. https://doi.org/10.3390/microorganisms11092233
Chicago/Turabian StyleJustiz-Vaillant, Angel A., Arlene Faye-Ann Williams-Persad, Rodolfo Arozarena-Fundora, Darren Gopaul, Sachin Soodeen, Odalis Asin-Milan, Reinand Thompson, Chandrashekhar Unakal, and Patrick Eberechi Akpaka. 2023. "Chronic Granulomatous Disease (CGD): Commonly Associated Pathogens, Diagnosis and Treatment" Microorganisms 11, no. 9: 2233. https://doi.org/10.3390/microorganisms11092233