
Genomic Characteristics and Functional Analysis of Brucella sp. Strain WY7 Isolated from Antarctic Krill
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolation and Transmission Electron Microscopy Observations
2.2. DNA Extraction, PCR Amplification and Phylogenetic Tree Construction
2.3. Genome Assembly
2.4. Genomic Bioinformatics Analysis
3. Results
3.1. Taxonomic Identification of 16S rDNA and Morphological Characteristics
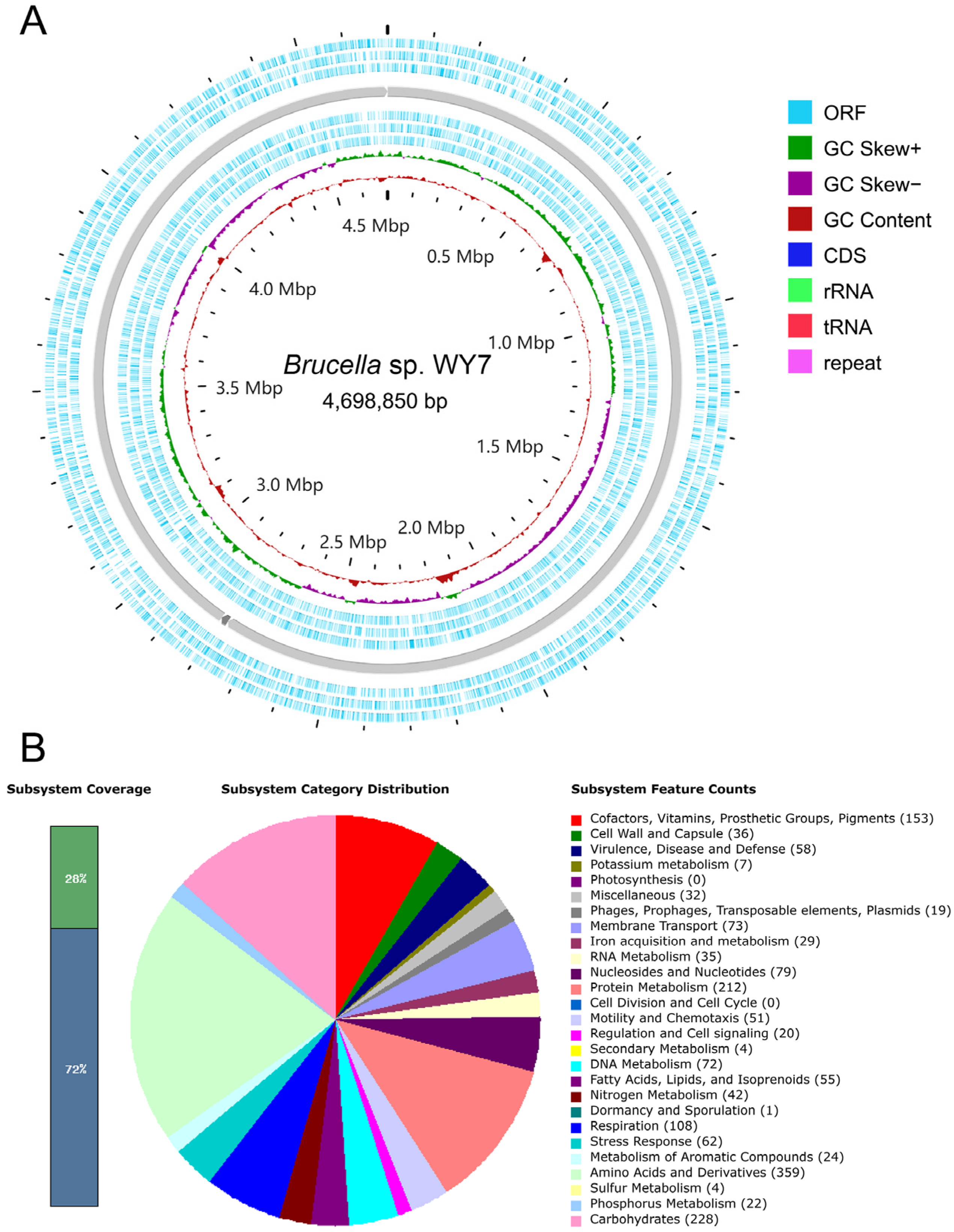
3.2. Genomic Characteristics
3.3. RAST Quick Genome Annotation
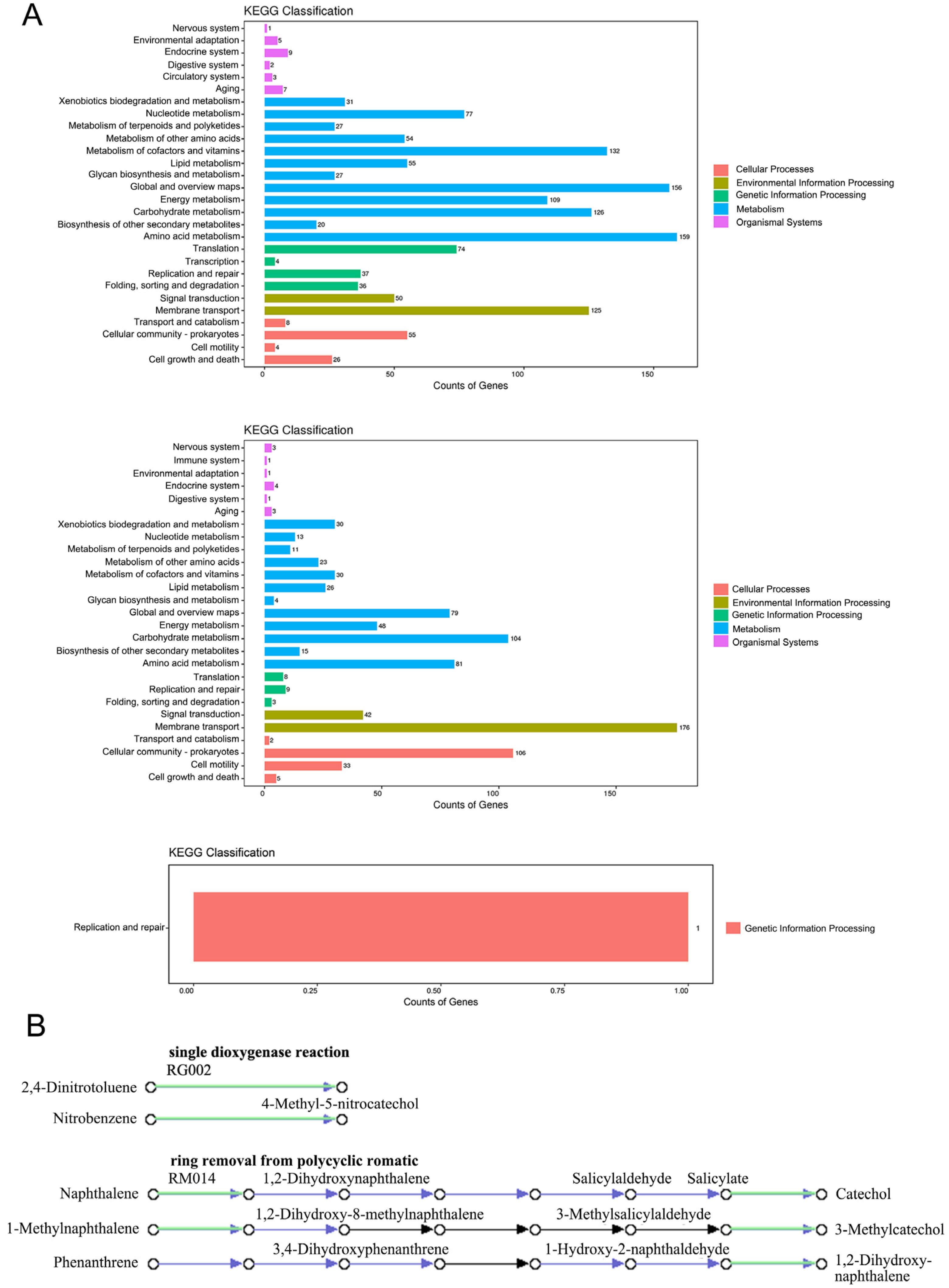
3.4. KEGG Database Annotation
3.5. CAZy Database Annotation
3.6. Genomic Islands Prediction
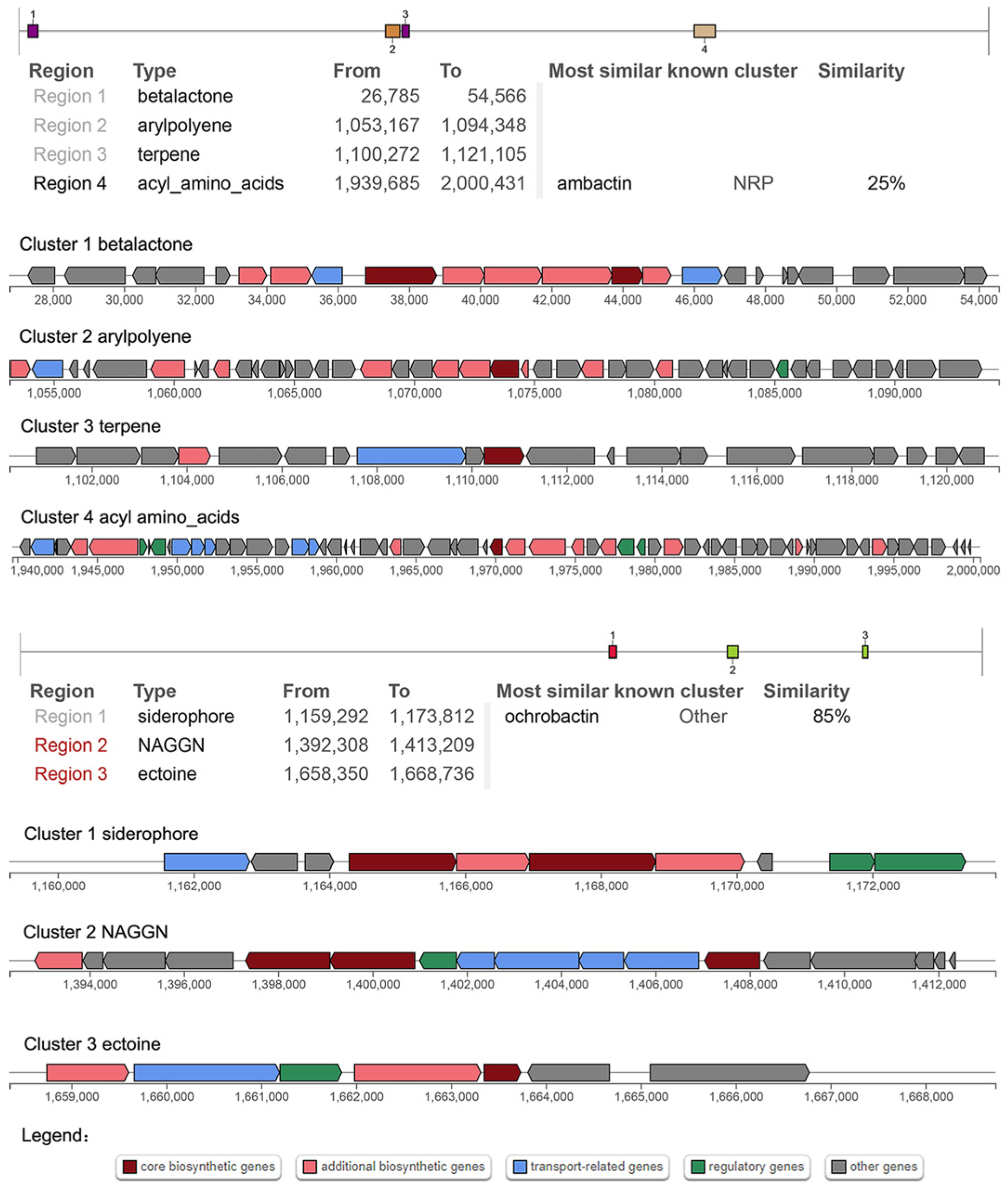
3.7. Secondary Metabolites
3.8. Pan-Genomic Comparative Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Meyer, B.; Kawaguchi, S. Antarctic marine life under pressure. Science 2022, 378, 230. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.J.; Ritson-Williams, R.; Sharp, K. Marine chemical ecology in benthic environments. Nat. Prod. Rep. 2011, 28, 345–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tian, X.; Yang, Q.; Lu, Y.; Ma, L.; Huang, H.; Fan, C. Diversity of secondary metabolites from two Antarctic microbes Rhodococcus sp. NJ-008 and Pseudomonas sp. NJ-011. Open J. Mar. Sci. 2014, 2014, 48424. [Google Scholar]
- Yang, Q. Taxonomic Identification and Bioactivity Screening of the Symbiotic Bacteria Strains of Euphausia superba from Antarctic Ocean. Appl. Mech. Mater. 2013, 295, 173–177. [Google Scholar] [CrossRef]
- Cui, X.; Zhu, G.; Liu, H.; Jiang, G.; Wang, Y.; Zhu, W. Diversity and function of the Antarctic krill microorganisms from Euphausia superba. Sci. Rep. 2016, 6, 36496. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, L.; He, J.; He, Z.; Wang, M.; Liu, Z.; Li, Z.; Wang, L.; Weng, S.; Guo, C.; et al. Environmental risk characteristics of bacterial antibiotic resistome in Antarctic krill. Ecotoxicol. Environ. Saf. 2022, 232, 113289. [Google Scholar] [CrossRef]
- Brinkmeyer, R.; Knittel, K.; Jürgens, J.; Weyland, H.; Amann, R.; Helmke, E. Diversity and structure of bacterial communities in Arctic versus Antarctic pack ice. Appl. Environ. Microbiol. 2003, 69, 6610–6619. [Google Scholar] [CrossRef]
- Chain, P.S.; Lang, D.M.; Comerci, D.J.; Malfatti, S.A.; Vergez, L.M.; Shin, M.; Ugalde, R.A.; Garcia, E.; Tolmasky, M.E. Genome of Ochrobactrum anthropi ATCC 49188 T, a versatile opportunistic pathogen and symbiont of several eukaryotic hosts. J. Bacteriol. 2011, 193, 4274–4275. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.S.; Watters, G.M. Comparing feedback and spatial approaches to advance ecosystem-based fisheries management in a changing Antarctic. PLoS ONE 2020, 15, e0231954. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, X.; Yu, Y.; Miao, J.; Leng, K.; Gao, H. Preparation process optimization, structural characterization and in vitro digestion stability analysis of Antarctic krill (Euphausia superba) peptides-zinc chelate. Food Chem. 2021, 340, 128056. [Google Scholar] [CrossRef]
- Holmes, B.; Popoff, M.; Kiredjian, M.; Kersters, K. Ochrobactrum anthropi gen. nov., sp. nov. from human clinical specimens and previously known as Group Vd. Int. J. Syst. Bacteriol. 1988, 38, 406–416. [Google Scholar] [CrossRef]
- Al-Mur, B.A.; Pugazhendi, A.; Jamal, M.T. Application of integrated extremophilic (halo-alkalo-thermophilic) bacterial consortium in the degradation of petroleum hydrocarbons and treatment of petroleum refinery wastewater under extreme condition. J. Hazard. Mater. 2021, 413, 125351. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Yu, J.; Chen, C. Isolation and identification of marine petroleum degradable bacterium AH07 and its degradation performance characteristics. J. Microbiol. 2020, 40, 32–36. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, C.; Wang, L.; Chairoungdua, A.; Piyachaturawat, P.; Fu, P.; Zhu, W. New ansamycins from the deep-sea-derived bacterium Ochrobactrum sp. OUCMDZ-2164. Mar. Drugs 2018, 16, 282. [Google Scholar] [CrossRef] [PubMed]
- Domingues, P.M.; Oliveira, V.; Serafim, L.S.; Gomes NC, M.; Cunha, Â. Biosurfactant production in sub-oxic conditions detected in hydrocarbon-degrading isolates from marine and estuarine sediments. Int. J. Environ. Res. Public Health 2020, 17, 1746. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.; Panda, A.N.; Kumari, K.; Adhya, T.K.; Raina, V. Comparative genomics of a paddy field bacterial isolate Ochrobactrum sp. CPD-03: Analysis of chlorpyrifos degradation potential. Indian J. Microbiol. 2020, 60, 325–333. [Google Scholar] [CrossRef]
- Wang, X.; Jin, D.; Zhou, L.; Zhang, Z. Draft genome sequence of Ochrobactrum anthropi strain W13P3, a halotolerant polycyclic aromatic hydrocarbon-degrading bacterium. Genome Announc. 2015, 3, e00867-15. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Fexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Felsenstein, J. Confdence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef]
- Hunt, M.; De Silva, N.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identifcation. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R.; Oliver, G.F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the rapid annotation of microbial genomes using subsystems technology(RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39 (Suppl. 2), W316–W322. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2008, 37 (Suppl. 1), D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Andreu, V.P.; Santos EL, C.; Carratore, F.D.; Lee, S.Y.; Medema, M.H.; Weber, T. The antiSMASH database version 2: A comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2019, 47, D625–D630. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]
- Zurdo-Piñeiro, J.L.; Rivas, R.; Trujillo, M.E.; Vizcaíno, N.; Carrasco, J.A.; Chamber, M.; Palomares, A.; Mateos, P.F.; Martínez-Molina, E.; Velázquez, E. Ochrobactrum cytisi sp. nov., isolated from nodules of Cytisus scoparius in Spain. Int. J. Syst. Evol. Microbiol. 2007, 57 Pt 4, 784–788. [Google Scholar] [CrossRef]
- Martin, J.D.; Ito, Y.; Homann, V.; Haygood, M.G.; Butler, A. Structure and membrane affinity of new amphiphilic siderophores produced by Ochrobactrum sp. SP18. JBIC J. Biol. Inorg. Chem. 2006, 11, 633–641. [Google Scholar] [CrossRef]
- Bhatia, M.; Sharp, M.; Foght, J. Distinct bacterial communities exist beneath a high Arctic polythermal glacier. Appl. Environ. Microbiol. 2006, 72, 5838–5845. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Seo, J.-S.; Keum, Y.-S.; Li, Q.X. Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 2009, 6, 278–309. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, B.; Ma, L.; Li, Y.; Cai, B. Nucleotide seouence analysis and enzyme characterization of the Pseudomonas sp. nd6 salicylate hydroxylase gene (nahG). Acta Sci. Nat. Univ. Nankaiensis 2004, 37, 95–99. [Google Scholar]
- Zhang, L. The Study of the Enzymatic Properties about Protocatechuate 3,4-Dioxygenase and Its Immobilization; East China University of Science and Technology: Shanghai, China, 2017. [Google Scholar]
- Zhang, X.; Zhu, J.; Cai, Z.; Zhou, J. The research advance of siderophores in marine microbes. Prog. Biochem. Biophys. 2022, 49, 1658–1671. [Google Scholar] [CrossRef]
- Shi, Q.; Li, L.; Huo, C.; Zhang, M.; Wang, Y. Research outline of marine natural products. Chin. Tradit. Herb. Drugs 2010, 41, 1031–1047. [Google Scholar]
- Zhang, S.; Hu, M.; He, Y.; Dong, Z. Research progress in microbial production and application of ectoines. Acta Microbiol. Sin. 2021, 61, 2250–2263. [Google Scholar] [CrossRef]
- Li, H.; Zhou, L.; Wang, Y.; Top, E.M.; Zhang, Y.; Xu, H. Degradative mobile genetic elements (MGEs) and their potential use in MGE-mediated biodegradation. Chin. J. Appl. Ecol. 2011, 22, 526–536. [Google Scholar]
- Dong, N.; Liu, D.; Fu, Y.; Yi, Z. Amplification and bioinformatics analysis of the higB gene from Mycobacterium tuberculosis. J. Parasit. Biol. 2016, 11, 505–508. [Google Scholar] [CrossRef]
- Lin, S.; Wang, X.; Chen, R. Analysis and identification of seven pairs of toxin-antitoxin systems in two cyanobacterial species. J. Trop. Oceanogr. 2022, 41, 119–134. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, L.; He, J.; Liu, Z.; Weng, S.; Wang, L.; He, J.; Guo, C. Whole genome sequencing and comparative genomic analyses of Planococcus alpniumensis MSAK28401T, a new species isolated from Antarctic krill. BMC Microbiol. 2021, 21, 288. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Chr.1 | Chr.2 | Plas. | Total |
|---|---|---|---|---|
| Genome size (Mb) | 2.79 | 1.89 | 0.019 | 4.70 |
| DNA coding region (Mb) | 2.44 | 1.70 | 0.017 | 4.16 |
| DNA G + C (%) | 57.61 | 56.57 | 55.17 | 57.18 |
| DNA scaffolds | 1 | 1 | 1 | 3 |
| Total genes | 2647 | 1718 | 24 | 4389 |
| RNA gene | 47 | 21 | 0 | 68 |
| Genes assigned to COGs | 2230 | 1432 | 7 | 3669 |
| Repeats | 150 | 79 | 0 | 229 |
| Subject Strain | Query Strain | dDDH (d4, in%) | C.I. (d4, in%) | The Origin of Subject Strain |
|---|---|---|---|---|
| Brucella anthropi ATCC 49188 (JAAVLS010000001.1) | WY7 | 94.8 | [93.2–96.1] | Type strain from human clinical specimens, blood cultures |
| Brucella anthropi NCTC 12168 (UGSA01000001.1) | WY7 | 94.8 | [93.2–96.1] | Type strain from human clinical specimens, blood cultures |
| Brucella lupini LUP21 (GCA 002252535.1) | WY7 | 82.5 | [79.7–85.1] | Plant root nodule samples |
| Brucella anthropi PBO (GCA 015326295.1) | WY7 | 78.8 | [75.9–81.5] | Plastic debris from sea coast, Qingdao, China |
| Brucella melitensis 16M (GCA 000007125.1) | WY7 | 25.0 | [22.7–27.5] | An infected goat |
| Species | WY7 | Brucella anthropi NCTC 12168 | Brucella anthropi ATCC 49188 | Brucella lupini LUP21 | Brucella anthropi PBO | Brucella melitensis 16M |
|---|---|---|---|---|---|---|
| WY7 | 100 | 99.07 | 99.07 | 97.49 | 97.21 | 80.55 |
| Brucella anthropi NCTC 12168 | 98.91 | 100 | 99.78 | 97.47 | 96.73 | 80.40 |
| Brucella anthropi ATCC 49188 | 98.70 | 99.77 | 100 | 97.26 | 96.55 | 80.15 |
| Brucella lupini LUP21 | 96.68 | 96.77 | 96.60 | 100 | 96.63 | 79.91 |
| Brucella anthropi PBO | 97.14 | 96.89 | 96.73 | 97.43 | 100 | 80.43 |
| Brucella melitensis 16M | 80.99 | 81.00 | 80.98 | 80.89 | 81.07 | 100 |
| Classification | Number of Genes | ||
|---|---|---|---|
| Chr.1 | Chr.2 | Plas. | |
| Cellular Processes | |||
| Cell growth and death | 26 | 5 | 0 |
| Cell motility | 4 | 33 | 0 |
| Cellular community—prokaryotes | 55 | 106 | 0 |
| Transport and catabolism | 8 | 2 | 0 |
| Environmental Information Processing | |||
| Membrane transport | 125 | 176 | 0 |
| Signal transduction | 50 | 42 | 0 |
| Genetic Information Processing | |||
| Folding, sorting and degradation | 36 | 3 | 0 |
| Replication and repair | 37 | 9 | 1 |
| Transcription | 4 | 0 | 0 |
| Translation | 74 | 8 | 0 |
| Metabolism | |||
| Amino acid metabolism | 159 | 81 | 0 |
| Biosynthesis of other secondary metabolites | 20 | 15 | 0 |
| Carbohydrate metabolism | 126 | 104 | 0 |
| Energy metabolism | 109 | 48 | 0 |
| Global and overview maps | 156 | 79 | 0 |
| Glycan biosynthesis and metabolism | 27 | 4 | 0 |
| Lipid metabolism | 55 | 26 | 0 |
| Metabolism of cofactors and vitamins | 132 | 30 | 0 |
| Metabolism of other amino acids | 54 | 23 | 0 |
| Metabolism of terpenoids and polyketides | 27 | 11 | 0 |
| Nucleotide metabolism | 77 | 13 | 0 |
| Xenobiotics biodegradation and metabolism | 31 | 30 | 0 |
| Organismal Systems | |||
| Aging | 7 | 3 | 0 |
| Circulatory system | 3 | 0 | 0 |
| Digestive system | 2 | 1 | 0 |
| Endocrine system | 9 | 4 | 0 |
| Environmental adaptation | 5 | 1 | 0 |
| Immune system | 0 | 1 | 0 |
| Nervous system | 1 | 3 | 0 |
| Total | 1419 | 861 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Z.; Wang, Y.; Ma, L.; Huang, S.; Wang, L.; He, J.; Guo, C. Genomic Characteristics and Functional Analysis of Brucella sp. Strain WY7 Isolated from Antarctic Krill. Microorganisms 2023, 11, 2281. https://doi.org/10.3390/microorganisms11092281
Feng Z, Wang Y, Ma L, Huang S, Wang L, He J, Guo C. Genomic Characteristics and Functional Analysis of Brucella sp. Strain WY7 Isolated from Antarctic Krill. Microorganisms. 2023; 11(9):2281. https://doi.org/10.3390/microorganisms11092281
Chicago/Turabian StyleFeng, Zhengqi, Yuanyuan Wang, Lingbo Ma, Shanzi Huang, Lumin Wang, Jianguo He, and Changjun Guo. 2023. "Genomic Characteristics and Functional Analysis of Brucella sp. Strain WY7 Isolated from Antarctic Krill" Microorganisms 11, no. 9: 2281. https://doi.org/10.3390/microorganisms11092281