
Freshwater Sponges as a Neglected Reservoir of Bacterial Biodiversity



1
Institute of Polar Sciences, National Research Council (CNR.ISP), Spianata S. Raineri 86, 98122 Messina, Italy
2
Zoological Station “Anton Dohrn”, Department of Ecosustainable Marine Biotechnology, Villa Pace, Contrada Porticatello, 98168 Messina, Italy
*
Author to whom correspondence should be addressed.
Microorganisms 2024, 12(1), 25; https://doi.org/10.3390/microorganisms12010025
Submission received: 29 November 2023
/
Revised: 18 December 2023
/
Accepted: 20 December 2023
/
Published: 22 December 2023
(This article belongs to the Special Issue Latest Review Papers in Environmental Microbiology 2023)
Abstract
:Freshwater sponges (Spongillida: Demospongiae), including more than 240 described species, are globally distributed in continental waters (except for Antarctica), where they cover both natural and artificial surfaces. However, fragmentary studies have targeted their microbiome, making it difficult to test hypotheses about sponge-microbe specificity and metabolic relationships, along with the environmental factors playing key roles in structuring the associated microbial communities. To date, particular attention has been paid to sponges (family Lubomirskiidae) that are endemic to Lake Baikal. Few other freshwater sponge species (e.g., Ephydatia spp., Eunapius spp., and Spongilla lacustris), from lakes and rivers spanning from Europe to South and North America, have been targeted for microbiological studies. Representatives of the phyla Proteobacteria, Bacteroidetes, and Actinobacteria largely predominated, and high differences were reported between the microbiome of freshwater and marine sponges. Several bacterial strains isolated from freshwater sponges can produce bioactive compounds, mainly showing antibiotic activities, with potential application in biotechnology. Understanding the roles played by sponge microbiomes in freshwater ecosystems is still in its infancy and has yet to be clarified to disentangle the ecological and evolutionary significance of these largely under-investigated microbial communities. This review was aimed at providing the main available information on the composition and biotechnological potential of prokaryotic communities associated with healthy freshwater sponges, as a neglected component of the global sponge microbiome, to stimulate researchers interested in the field.
1. Introduction
Among the oldest extant multicellular animals, sponges (Phylum Porifera) are sessile benthic filter-feeding macroinvertebrates commonly found in aquatic habitats worldwide. All sponges pump high amounts of water through their specialized aquiferous system. A collagenous extracellular matrix (called mesohyl) covers the system of sponge canals, filling the space between the ectoderm (i.e., the outer cell layer) and the pinacoderm (composed of cells lining the inner canals). Choanocytes (i.e., flagellated cells), organized in choanocyte chambers, are responsible for the flux of water from the exterior, through pores called ostia, to the internal canal system. Food particles suspended in pumped water are transferred into the mesohyl and then digested by specialized amoeboid cells, called archaeocytes, which are embedded in the three-dimensional extracellular matrix of the sponge [1].
The ecological, evolutionary, and microbiological significance of sponges is unquestioned [2]. Filtering large volumes of water, sponges play crucial roles in the coupling of benthic and pelagic ecosystems due to the recycling of both inorganic and organic nutrients (e.g., silicon, carbon, and nitrogen) [3,4]. Thanks to their complex three-dimensional structures, sponges provide habitat and refuge from predators for several species (including microinvertebrates, microalgae, and microeukaryotes), thus enhancing ecosystem biodiversity and functioning [5]. Sponges can be colonized by prokaryotic symbiotic assemblages, with most of them that are species-specific and phylogenetically diverse, strongly differing from those inhabiting the surrounding environment. It has been demonstrated that the body weight of some marine sponge species (namely, high microbial abundance sponges, HMA) can be composed of 40% of microorganisms [6,7], reaching an abundance of 108–1010 microbes g−1 sponge wet weight, contrary to low microbial abundance (LMA) sponges that host around 105–106 microbial cells g−1 [8,9]. Symbiotic microbes are highly involved in the ecology and physiology of their host (for instance, its development, behavior, and resistance to disease); for this reason, the sponge host and the associated microbiota are often altogether described as a “holobiont”, i.e., a unique biological entity with genetic features (i.e., the hologenome) that are also contributed by the microorganisms [10].
Freshwater sponges (Spongillida: Demospongiae) include more than 240 described species (organized into six families and 47 genera) [11]. In inland waters, both lentic and lotic systems (e.g., lakes, ponds, rivers, streams, man-made reservoirs, and garden ponds) can be found covering both natural (e.g., stones, wood, macrophytes, or invertebrates) and anthropic (e.g., glass, metal) substrata assuming branched, encrusting, or cloddy bulk shapes. They largely varied in color and dimensions, ranging from a few mm to 1.50 m in size [12]. A greenish appearance is frequent in light-exposed habitats, due to the occurrence of algal or cyanobacterial symbionts supplying photosynthates to the sponge and intervening in the protection against UV radiation [1,13]. Freshwater sponges are highly adapted to drastic changes characterizing environmental conditions of freshwater systems (e.g., thermal fluctuations, fluctuating water levels, long-lasting desiccation, temporary anoxic conditions, and pollutant occurrence) thanks to modifications at the molecular, physiological, and structural level [11]. For instance, freshwater sponges can produce gemmules, encapsulating undifferentiated cells into silica structures, to survive under winter conditions [11].
To date, several studies have examined microorganisms associated with marine Porifera, which include more than 95% of known sponge species. However, even if freshwater sponges offer similar ecosystem functions in freshwater habitats [2], sporadic and fragmentary studies have targeted their microbiology, making it difficult to test hypotheses about sponge-microbe specificity (even in unconnected freshwater systems) and metabolic relationships, along with the environmental factors playing key roles in structuring the associated microbiome [2]. Particular attention has been paid to the sponge species Lubomirskia baicalensis, including diseased individuals, which are endemic to Lake Baikal [14,15,16,17,18]. Conversely, fragmentary data are reported on the microbiota associated with a few other sponge species (e.g., Corvospongilla lapidosa, Ephydatia fluviatilis, E. muelleri, Eunapius carteri, Spongilla lacustris, and Tubella variabilis), from other freshwater systems (Figure 1).
Most studies applied culture-dependent methods for the characterization of bacterial isolates, including their potential as producers of biotechnological relevant molecules [16,17,18,19], whereas high-resolution taxonomic data on the whole communities, obtained by next-generation sequencing technologies, remain still limited (e.g., [2,20,21,22,23]). Overall, the prokaryotic communities are generally characterized by the predominance of Proteobacteria, Bacteroidetes, and Actinobacteria and highly differ from those determined in marine sponges [21]. Sugden et al. [2] reported for the first time the comparison between sponge-associated communities and the surrounding environments (e.g., water and biofilm). This latter aspect is of major importance to establish if discrepancies between prokaryotic communities in sponges and water can match the differences that may be observed between benthic and planktonic communities rather than the functional significance of host-microbe specificity [2].
The sponge immune system constitutes a first line of defense against transient microbiota, including the production of biomolecules that recognize conserved microbial structures [24]. Moreover, bacterial symbionts are probably involved in the sponge defense strategies to prevent predation, e.g., by fishes and mollusks, and impede colonization by unwanted organisms (e.g., microbial biofilms and fouling) through the production of antagonistic metabolites [25]. Different from marine sponges, our current knowledge on this aspect in inland water sponges remains poorly investigated. It is noteworthy that more than 100 distinct bioactive compounds, that can be useful for humans, have been detected in freshwater sponges and some of them might originate from symbiotic bacteria [26,27].
This review was aimed at providing the main available information on the composition and biotechnological potential of prokaryotic communities associated with healthy freshwater sponges, as a neglected component of the global sponge microbiome.
2. Prokaryotic Communities Associated with Freshwater Sponges: Main Findings by Culture-Independent Approaches
In the following sections, our current knowledge on the composition, and possibly functions, of bacterial communities associated with freshwater sponges, as determined by culture-independent methods (including clone library construction and next-generation sequencing, NGS), is showcased per sponge species (listed in Table 1).
2.1. Ephydatia spp.
E. fluviatilis (Linnaeus, 1759) is a sponge species common to continental water bodies (both lotic and lentic) in the northern hemisphere, particularly in Central Europe. The molecular diversity and composition of bacteria in E. fluviatilis, from the artificial lake Vinkeveense Plassen (Utrecht, The Netherlands), was explored for the first time by Costa et al. [28], who suggested a selective process by the host organism to choose associated microbes. By applying the polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) fingerprints, the authors observed that sponge- and water-derived bacterial communities differed in composition at the phylum level, with no or negligible overlaps between the E. fluviatilis and water-derived phylotypes within bacterial taxa. For instance, Actinobacteria, Proteobacteria, and Bacteroidetes dominated in the freshwater clone library, whereas Proteobacteria, Planctomycetes, Actinobacteria, Bacteroidetes, Chlamydiae, and Verrucomicrobiota were more abundant in water. The co-dominance of other bacterial phyla, such as the candidate phylum TM7, was also reported. Interestingly, a distinct and less diversified actinobacterial community was observed in E. fluviatilis, with a sharp selection of an uncultured actinobacterial phylotype in the order Acidimicrobiales [28]. Within Gammaproteobacteria, a previously unsuspected complexity of E. fluviatilis-associated Pseudomonas assemblages was revealed by the search for the gacA gene as a phylogenetic marker alternative to conventional 16S rRNA marker gene, which instead failed to reflect the multiplicity of these organisms in their sponge host [25]. Within Alphaproteobacteria, two clusters recovered from E. fluviatilis were related (at about 93–94% sequence similarity) to sequences previously obtained from S. lacustris [1] and L. baicalensis [29]. The same was observed for a sponge-specific cluster within Bacteroidetes, which was previously reported within L. baicalensis community [29], and for a Polynucleobacter sp. cluster that was similar (>96%) to a Polynucleobacter necessarius from S. lacustris [1]. According to the authors, whether these phylotypes represent freshwater sponge-specific lineages, thus supporting the hypothesis on the existence of co-evolutionary relationships between hosts and symbionts, remains to be demonstrated.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Studies targeting freshwater sponge prokaryotic communities by culture-independent methods.
Table 1.
Studies targeting freshwater sponge prokaryotic communities by culture-independent methods.
| Sponge Species | Freshwater Sampling Site | Reference(s) |
|---|---|---|
| Baikalospongia sp. | Lake Baikal (Russia) | [30,31] |
| Baikalospongia bacillifera (Dybowsky, 1880) | Lake Baikal (Russia) | [22] |
| Baikalospongia intermedia (Dybowski, 1880) | Lake Baikal (Russia) | [32] |
| Corvospongilla lapidosa (Annandale, 1908) | Talegaon Dabhade and Pashan (India) | [20] |
| Ephydatia fluviatilis (Linnaeus, 1759) | Vinkeveense Plassen Lake (The Netherlands) | [25,28] |
| Ephydatia muelleri (Lieberkühn, 1856) | Six locations in the Northern Hemisphere | [22] |
| Sooke, Cowichan, and Nanaimo Rivers (Canada) | [2] | |
| Eunapius carteri (Bowerbank, 1863) | Talegaon Dabhade and Pashan (India) | [20] |
| Lubomirskia baicalensis (Pallas, 1776) | Lake Baikal (Russia) | [15,30,31,32,33] |
| L. abietina (Swartschewsky, 1901) | Lake Baikal (Russia) | [33] |
| Spongilla lacustris (Linnaeus, 1759) | Lake Staffelsee (Germany) | [1] |
| Pichlinger See Lake (Upper Austria) | [23] | |
| Swartschewskia papyracea (Dybowsky, 1880) | Lake Baikal (Russia) | [32] |
| Tubella variabilis (Bonetto and Ezcurra de Drago, 1973) | Artificial channel (Brazil) | [21] |
E. muelleri (Lieberkühn, 1856) is a cosmopolitan and globally abundant sponge species. Its chromosomal-level genome has been well annotated [22]. This species is frequently studied for some physiological, biological, and genetic sponge aspects, especially in relation to animal evolution [34,35]. Some sets of E. muelleri genes have been proven to be involved in the establishment and maintenance of symbiotic interactions [36]. To date, this sponge has been the subject of two papers dealing with its associated microbiomes, analyzed by culture-independent approaches [2,22]. Kenny et al. [22] reported a microbiome composition of E. muelleri (from six different freshwater locations of the Northern Hemisphere) that was comparable to that observed in most marine demosponges, with Proteobacteria and Bacteroidetes as predominant microbial components. However, differently from marine sponges, E. muelleri hosted a large fraction of Betaproteobacteriales (absent in marine sponges). Overall, microbial content differed by geographic location. For instance, samples from the Sooke Reservoir had a higher abundance of Firmicutes and Campylobacteria, whereas those from Maine were characterized by a moderate abundance of Cyanobacteria. Notably, despite that sponge individuals were collected from distant sites (with potentially different ecological features), the authors detected few amplicon sequence variants (ASVs) that were shared among all samples, occurring at different relative percentages, including Burkholderiaceae (order Betaproteobacteriales) and Ferruginibacter (order Chitinophagales). These findings represented a baseline for the study of species-specific patterns of host–microbe association in freshwater systems at a broader scale. Later, Sugden et al. [2] applied the 16S rRNA gene amplicon sequencing complemented with shotgun metagenomics to describe the microbiome of E. muelleri from the Sooke, Nanaimo, and Cowichan Rivers on Vancouver Island (British Columbia, Canada), also in relation to ambient water and adjacent biofilm. The authors demonstrated that E. muelleri core microbiome, besides being different from those of the surrounding environment, included Comamonas, Diaphorobacter, Methylotenera, Rhodoferax, unclassified Rhodospirillales, and Sediminibacterium, with the predominance of Sediminibacterium, Comamonas and Rhodospirillales (overall accounting for 58%). Notably, the dominant Sediminibacterium (as amplicon sequence variant, ASV) in sponge communities differed from the dominant Sediminibacterium ASV in both water and biofilm samples. Conversely, the most abundant ASV for Comamonas and Rhodospirillales were consistent across all tested matrices and rivers. Interestingly, most abundant Sediminibacterium and Rhodospirillales sequences were closely related (sequence identities ≥ 98.42%) to uncultured bacteria from different sponge species from a number of freshwater systems [1,14,15,16,17,18,20,28], supporting the hypothesis that evolutionarily conserved sponge-bacteria associations may exist also in freshwater environments. Although the microbiome composition was largely conserved among different rivers, as was previously observed by Kenny et al. [22], sponges were distinguished by their origin based on a few microbial features. In detail, higher relative abundances were determined for Pseudarcicella, Flavobacterium, and Fluviicola in Sooke sponges; Polynucleobacter in Nanaimo sponges; and Sediminibacterium and Parcubacteria in Cowichan sponges. This finding again suggested that other environmental or host-specific variables may affect the observed geographic variations. Results obtained by Sugden et al. [2] differed from those previously obtained by Kenny et al. [22] for E. muelleri sponges and gemmules from upstream of the Sooke River, with the microbiome that better resembled those of analyzed biofilms. Finally, shotgun metagenomes and metagenome-assembled genomes revealed that the microbiome of E. muelleri can show some compositional and functional similarities (e.g., defense-related proteins and genes for vitamin B12 production) with prokaryotic communities associated with marine sponges [2].
2.2. Eunapius carteri and Corvospongilla lapidosa
The microbiota of the globular sponge E. carteri (Bowerbank, 1863) and the encrusting sponge C. lapidosa (Annandale, 1908) (from the permanent freshwater lake located at Talegaon Dabhade and Pashan, India, respectively) was explored for the first time using next generation sequencing (NGS) technology by Gaikward et al. [20]. The authors also analyzed lake water for microbiota composition. Overall, 14 bacterial phyla were detected, with more than 2900 and 980 OTUs (higher than those generally reported for marine sponges) that were obtained from C. lapidosa and E. carteri, respectively. The two sponge-associated microbial communities strongly differed: E. carteri community was dominated by Firmicutes, followed by Proteobacteria and Cyanobacteria; C. lapidosa community was characterized by a higher abundance of Proteobacteria, followed by Planctomycetes, Cyanobacteria, and Actinobacteria. The authors suggested that this finding probably was dependent on the differences encountered in the measured environmental variables, highlighting the crucial role played by the habitat in structuring the associated microbial communities. Among the main phyla, Cyanobacteria are considered minor components of the bacterial communities associated with freshwater sponges. However, Gaikward et al. [20] determined a high abundance of Cyanobacteria in both sponges (comparable with those reported for marine sponges). In particular, Synechococcus sequences were abundantly present only in E. carteri, whereas Planktothrix and Planktothricoides were abundant in C. lapidosa. Nevertheless, the role played by Cyanobacteria in their association with sponges (as a food or symbiont) remains to be elucidated. The study also revealed that the structure of both microbial communities was significantly different from their respective water samples (i.e., dominant Proteobacteria, followed by Bacteroidetes, Actinobacteria, and Cyanobacteria in the water next to E. carteri; dominant Actinobacteria, followed by Bacteroidetes, Proteobacteria, Planctomycetes, and Cyanobacteria in the water next to C. lapidosa), implying that the host genetic factors could be also involved in the species-specific association. Notably, Nitrospirae, Chloroflexi, Chlamydiae, and Acidobacteria occurred only within the C. lapidosa bacterial community. Conversely, Firmicutes were retrieved only in association with E. carteri, with more than 50% of their sequences being affiliated with the genus Clostridium (absent in the surrounding water). Gaikward et al. [20] supposed that the high abundance of Clostridium might be due to the ability of members in this genus to utilize some complex molecules (such as proteoglycans, glycoproteins, collagen, and spongin) that are present in the sponge extracellular matrix. However, the authors deserved to verify if, as observed in marine sponges, the occurrence of Clostridium spp. might derive from the exposition to environmental stresses (e.g., toxic chemicals). Finally, based on the detection of several bacterial lineages (belonging to Firmicutes, Actinobacteria, Proteobacteria, and Planctomycetes) that are known to produce compounds of biotechnological relevance, the authors encouraged the isolation and characterization of microbes from freshwater sponges to be screened for their biotechnological potentialities.
2.3. Lubomirskiidae Family from the Lake Baikal
At least 14 species of freshwater sponges are endemic to Lake Baikal (Russia) and belong to the Lubomirskiidae family. The Lubomirskiidae include species such as Lubomirskia baicalensis, Baikalospongia bacillifera, B. intermedia, B. martinsoni, and Swartschewskia papyracea. Most studies on the symbiotic bacterial communities have been focused on L. baicalensis and Baikalospongia spp., as reported below.
L. baicalensis (Pallas, 1776) is the dominant endemic sponge species of Lake Baikal. It lives in symbiosis with a green okadaic acid-producing dinoflagellate (closely related to related to Gymnodinium sanguineum), involved in the host survival in the iced lake in winter [37]. The diversity of the microbial community associated with the branching L. baicalensis, together with that of an encrusting Baikalospongia sp., was evaluated by NGS for the first time by Gladkikh et al. [30], highlighting a complexity in diversity level that was comparable to that estimated for marine sponges. Overall, in L. baicalensis 6873 (out of 7071) 16S rRNA gene sequences belonged to the bacteria domain (with 2935 of them that were unique). A total of 426 identified phylotypes grouped in four main bacterial phyla, i.e., Bacteroidetes (48% of total sequences), Proteobacteria (28%), Actinobacteria (14.7%), and Planctomycetes (7.3%), followed at lower abundance by Verrucomicrobiota, Nitrospirae, OD1, and Chloroflexi. Sequences not affiliated with known phyla were also observed. These findings were in line with previous observations previously made by applying clone library approach, allowing the detection of Actinobacteria (37%), Proteobacteria (Alpha- and Betaproteobacteria, 22 and 13%, respectively), Verrucomicrobia (11%), Bacteroidetes (7.5%), Cyanobacteria (7.5%), and Nitrospira (2%), even if they occurred at different relative percentages [15] As was observed for Baikalospongia sp. from the same study by Gladkikh et al. [30], Sediminibacterium affiliates predominated, accounting for 40.4% of the total community. Among Proteobacteria, Polynucleobacter necessarius (family Burkholderiaceae, Betaproteobacteria) constituted 22.7% of the total community, in accordance with results on clones from E. fluviatilis [28] and Baikalospongia sp. [30]. Additionally, Pedomicrobium australicum (family Hyphomicrobiaceae; Alphaproteobacteria) represented 4.9% of the bacterial community associated with L. baicalensis. The third most abundant phylum, i.e., Actinobacteria, was exclusively composed of the common planktonic species Planktophila limnetica. Overall, 6817 (out of 7042) 16S rRNA gene sequences from Baikalospongia sp. belonged to the bacteria domain (with 2601 of them that were unique) [30]. A total of 428 phylotypes were identified and grouped into four main bacterial phyla, i.e., Bacteroidetes (52.3% of total sequences), Proteobacteria (28.7%), Actinobacteria (9.5%), and Planctomycetes (7.7%), followed at lower abundance by Verrucomicrobiota, OD1 and Chloroflexi. Sequences not affiliated with known phyla were also observed. Within Bacteroidetes, the genus Sediminibacterium accounted for 47.0% of the total community. Among Proteobacteria, Polynucleobacter necessarius (family Burkholderiaceae, Betaproteobacteria) constituted 12.0% of the total community, in accordance with previous results on clones from E. fluviatilis [28]. The third most abundant phylum, i.e., Actinobacteria, was exclusively composed of the common planktonic species Planktophila limnetica. Finally, Planctomycetes were mostly represented by Phycisphaera mikurensi (7.2% of the total community). A high sequence homology (95–97%) was observed between the two analyzed sponge species, namely Baikalospongia sp. and L. baicalensis, and the bacterioplankton from the same site of collection in Lake Baikal. Furthermore, sponge communities shared 75 common OTUs at the level of species (97% homology), with over 50% of the bacterial phylotypes of each sponge being unique, and 55% of the planktonic phylotypes that were not retrieved in associations with sponges.
Jung et al. [31] tested a new isolation method from L. baicalensis and Baicalospongia sp., comparing the obtained results with those from pyrosequencing. Overall, 13,964 and 7496 reads were analyzed from L. baicalensis and Baicalospongia sp., respectively. Actinobacteria, Alphaproteobacteria, Firmicutes, Betaproteobacteria, Cyanobacteria, Planctomycetes, and Verrucomicrobia were retrieved in both sponges (except for Verrucomicrobia in L. baicalensis). Cyanobacteria predominate within both associated bacterial communities, accounting for 70–78% of total reads, whereas Gammaproteobacteria abundance was negligible (<1%).
Seo et al. [32] analyzed the bacterial diversity of the Baikalian sponges L. baicalensis, B. intermedia Dybowsky, 1880 and S. papyracea (Dybowsky, 1880) (the latter reported for the first time) by pyrosequencing highlighted some differences in their bacterial community composition, suggesting that bacterial symbiont community composition was mainly affected by environmental conditions or gradients across water depths in Lake Baikal. A total of 7496, 13,654, and 4941 reads were retrieved from B. intermedia, L. baicalensis, and S. papyracea, respectively. Overall, Cyanobacteria predominated (range 42–78%), followed by Proteobacteria. In particular, Cyanobacteria were affiliated with the orders Synechococcales, Chroococcales (found only in L. baicalensis and S. papyracea) and Nostocales (retrieved only in B. intermedia and S. papyracea). The abundance of the genus Prochlorococcus was in the range of 40–74% in all sponge species, with Prochlorococcus marinus surprisingly, being generally found in the marine environment, occurring only in S. papyracea. B. intermedia and L. baicalensis hosted a highly similar bacterial community, differing from that associated with S. papyracea (showing a higher bacterial diversity). Actinobacteria abundance was three times higher in S. papyracea (13.4%) than B. intermedia and L. baicalensis (3.87 and 3.46%, respectively), while Acidobacteria and Gemmatimonadetes occurred only in S. papyracea. Ten reads in the orders Micrococcales, Bacteroidales, Verrucomicrobiales, and Solirubrobacterales occurred only in S. papyracea. The authors hypothesized that the bacterial community associated with S. papyracea was sponge-specific and adapted for specific environmental conditions (such as lower light levels or higher pressure than the other two sponge species).
More recently, Kenny et al. [33] compared the genomic content and microbiome of L. baicalensis and B. bacillifera, in addition to L. abietina Swartschewsky, 1901, to gain knowledge on their molecular evolution to adapt to Lake Baikal’s unique environment. Overall, the dataset included a small number of bacterial sequences, representing a subsample of complete bacterial diversity. According to the authors, bacterial genomes were not inordinately represented in the dataset, indicating that they were not well assembled. However, the obtained results were generally consistent with previous data on other freshwater sponge species (e.g., [1,20,28,32]). Briefly, in L. baicalensis Actinobacterial sequences (1589 annotated contigs) were predominant, followed by Proteobacteria (1087 contigs) and Candidatus Tectomicrobia sp. (191 contigs). Firmicutes, Bacteroidetes, Cyanobacteria, Verrucomicrobia, and Chlamydiae (in order of decreasing occurrence) were less represented. In L. abietina 407 and 203 proteobacterial and Candidatus Tectomicrobia contigs were retrieved, followed by Actinobacteria, Firmicutes, Bacteroidetes, Cyanobacteria, Verrucomicrobia, and Chloroflexi. Chlamydiae contigs were only eight. In B. bacillifera, Kenny et al. [33] observed the predominance of Proteobacteria (392 contigs), followed by Candidatus Tectomicrobia (157 contigs) and, at a lesser extent, by Bacteroidetes, Firmicutes, Actinobacteria, Cyanobacteria, and Verrucomicrobia. Only nine contigs of the obligately intracellular Chlamydiae occurred. Overall, the most common bacterial symbiont in the three sponge species was Candidatus Entotheonella geminae. According to the authors, this symbiotic bacterium may be necessary for the survival of the host playing roles that are useful in freshwater in general, and Lake Baikal in particular, such as the protection against heavy metals, supply of energy in anaerobic conditions, and support in CO2 fixation.
2.4. Spongilla lacustris
S. lacustris (Linneus, 1759) is a widespread (and seldom fast-growing, especially in Central Europe) sponge species generally inhabiting temperate regions of the Northern Hemisphere. Although it is a very effective biological filter, being able to remove and consume large amounts of particulate matter in water, S. lacustris growth mainly depends on photosynthetic products supplied by the intracellular symbiotic algae [38,39]. To the best of our knowledge, to date, the bacterial community of S. lacustris has been described in depth only in a couple of papers by applying culture-independent methods [1,23]. Gernert et al. [1] constructed 16S rRNA gene libraries from both sponge tissues and Lake Staffelsee (Germany) water. By the application of the restriction fragment length polymorphism (RFLP) analysis performed on 9190 freshwater sponge-derived clones, 45 clones (clustering in six major restriction patterns) were selected for sequencing. The authors observed the predominance of Alphaproteobacteria and Actinobacteria (51 and 36%, respectively), followed by Betaproteobacteria and Chloroflexi (5% each). The sponge-associated bacterial community was highly similar to that observed for Lake Staffelsee. However, two alphaproteobacterial sequences appeared to be novel and freshwater sponge-specific.
More recently, Graffius et al. [23] investigated the microbiome of S. lacustris from Lake Pichlinger See (Upper Austria) using a combined approach, which coupled the analysis of bacterial isolates (including their genome analysis) with those of 16S rRNA gene amplicons and metagenomes from sponge tissue DNA extracts. As it was determined by 16S rRNA gene sequencing, a wide fraction of ASVs was not classified at the genus level, being strongly related to uncultured and unclassified Bacteroidetes, Alphaproteobacteria, Gammaproteobacteria, and Betaproteobacteria, highlighting a high degree of phylogenetic novelty in the S. lacustris microbiome. Metagenome binning, resulting in 20 metagenome-assembled genomes (MAGs) did not reveal the occurrence of sponge-related taxa. Instead, MAGs are mostly related to metagenomes of freshwater and marine origin, as well as hot springs, hypersaline lakes, and pathogenic isolates from shrimps and diseased fish.
2.5. Tubella variabilis
T. variabilis (Bonetto and Ezcurra de Drago, 1973) is an encrusting thin sponge species. It is beige and green in color and fragile to moderately soft in consistency [40]. Laport et al. [21] compared the microbial community structure of T. variabilis specimens from an artificial channel, providing water to fish farm tanks (Recife, Pernambuco, Brazil), with that of the surrounding freshwater. Bacterial diversity was higher in sponges than in water. Overall, Proteobacteria dominated both communities, followed in their relative abundances by Bacteroidetes, Acidobacteriota, Verrucomicrobiota, and Cyanobacteria. However, Alphaproteobacteria were enriched in T. variabilis, whereas Betaproteobacteria dominated in freshwater. Among Bacteroidetes, which were more abundant in the sponge than in water, Cytophagia-related sequences were a hundred times higher in T. variabilis than in freshwater. To gain further insight, the authors tested the difference in the relative abundance of the most abundant OTUs (representing 64% of the total community). Among them, 20 strongly differed between the two habitats (sponge and water). For instance, the methanotrophic genus Methylosinus (among Alphaproteobacteria) and family Cytophagacea (among Bacteroidetes) were enriched in T. variabilis. Conversely, the genera Methylocaldum, Sulfuricurvum and genus C39 (order Rhodocyclales), and family Comamonadacea were more abundant in freshwater. Laport et al. [21] also compared the richness (measured by number of OTUs) and structure of the microbial community of T. variabilis with those of two other sponges, namely E. carteri and C. lapidosa from Indian freshwaters [20], along with 32 different sponges of marine origin. T. variabilis and C. lapidosa harbored the richest communities. Notably, freshwater and marine sponges are markedly grouped in separated clusters by nMDS. Since freshwater sponges were collected from geographically distant areas (i.e., India and Brazil) (collected in Brazil), the authors suggested that water salinity might be a possible main factor structuring the microbial community associated with sponges.
An overview of the freshwater sponge species investigated so far, and their geographical locations is given in Figure 2. It is a graphical representation of available data regarding the taxonomic composition of bacterial communities associated with freshwater sponges. Despite some bacterial taxa being overall shared between the various investigated sponge species, it is possible to appreciate the presence of some peculiar taxa in certain sponges, such as Spirochaetes, Tenericutes, and Nitrospirae.
3. Bacterial Isolates from Freshwater Sponges: Description and Biotechnological Potential
A number of bacterial strains have been isolated and described from freshwater sponges so far. In the following sections, current data on bacterial isolates, including the description of novel species, are reported per sponge species (cfr. Section 3.1), if dealing with the characterization of the cultivable bacterial fraction, or per biotechnological potential (cfr. Section 3.2), if dealing with isolates of biotechnological value.
3.1. Characterization of the Cultivable Fraction of the Freshwater Sponges-Associated Bacterial Communities
To date, the cultivable fraction of the bacterial communities associated with freshwater sponges has been analyzed for eight sponge species, spanning from Austria to Russia (Table 2).
3.1.1. Baikalospongia bacillifera, B. intermedia, Lubomirskia fusifera, L. baicalensis and Swartschewskia papyracea from Lake Baikal
A total of 77 bacterial strains were isolated from the sponges B. bacillifera Dybowski, 1880 (58 isolates), L. fusifera Soukatschoff, 1895 (13 isolates), and S. papyracea (Dybowsky, 1880) (6 isolates) collected in Lake Baikal near the village of Bol’shie Koty [14]. Strains affiliated with the genera Bacillus, Flavobacterium, Pseudomonas, and Acinetobacter were isolated from all sponge species. Micrococcus and Arthrobacter occurred only in L. fusifera and B. bacillifera, whereas Sarcina was retrieved only in S. papyracea. However, some genera were also isolated from lake water, with Pseudomonas that prevailed in water, and Bacillus, Micrococcus, and Sarcina that were more abundant in sponges. Parfenova et al. [14] isolated psychrophilic bacteria from the sponges L. baicalensis, B. bacillifera, and B. intermedia Dybowsky, 1880 collected near Cape Berezovyi (Lake Baikal). Streptomyces and Micromonospora affiliates were probably permanent components of the bacterial communities. In particular, Micromonospora accounted for 68, 69, and 90% of all actinomycetes in L. baicalensis, B. intermedia, and B. baicalensis, respectively. Later on, Jung et al. [31] applied a novel method, called I-tip, for the isolation of bacteria from L. baicalensis and Baicalospongia sp. The method missed only two major phyla detected by pyrosequencing, as it allowed the isolation of 34 bacterial species from five major phyla (i.e., Actinobacteria, Alphaproteobacteria, Betaproteobacteria, Firmicutes, and Gammaproteobacteria). Among them, Nocardia, Rhodococcus, Microbacterium, Brevundimonas, Sphingomonas, Acinetobacter, and Pseudomonas representatives were isolated from both sponge species. Conversely, three major phyla (namely, Betaproteobacteria, Firmicutes, and Gammaproteobacteria), including 16 bacterial species, were obtained by standard cultivation method, failing the detection of some major phyla determined by pyrosequencing. However, the I-tip-derived culture collection did not include a high abundance of Gammaproteobacteria, such as the commonly isolated Pseudomonas. Conversely, Actinobacteria and Alphaproteobacteria were enriched by the I-tip method. The authors conclude that the I-tip method can better represent the natural bacterial diversity, narrowing the gap between cultivated and uncultivated species, at least in the case pf host-associated microbial communities.
3.1.2. Ephydatia sp.
Based on the moderate relative abundance (14.4% of total bacterial abundance) of Planctomycetes reported by Costa et al. [28], Kohn et al. [41] enriched with N-acetyl-d-glucosamine (NAG) as sole carbon source samples of an Ephydatia sp. from Lake Constance (Germany), targeting bacteria from the phylum Planctomycetes. Following this approach, the authors isolated and first described a novel species, namely Planctopirus ephydatiae (strain spb1T). Contrary to chemotaxonomic measurements (including cellular fatty acid analysis) that showed similarities of strain spb1T with the related species P. limnophila (DSM 3776T), the genomic analysis revealed that strain spb1T represented a yet unknown species of Planctomycetes.
3.1.3. Eunapius fragilis
Two E. fragilis (Leidy, 1851) individuals were collected 1.5 km apart in the St. Lawrence River, North America [42]. A total of 851 isolates were obtained and identified as representatives of four major phyla commonly associated with sponges, i.e., Proteobacteria (including the genera Yersinia, Rahnella, Enterobacter, Pseudomonas, and Delftia), Actinobacteria (genera Micromonospora, Verrucosispora, and Streptomyces), Bacteroidetes (genus Chryseobacterium), and Firmicutes (genera Bacillus and Paenibacillus). The authors demonstrated that the two sponges shared many of the same genera but exhibited higher diversity at the species and possibly subspecies level. Isolates were screened for specialized metabolite production (see Section 3.2).
3.1.4. Spongilla lacustris
Graffius et al. [23] identified 197 bacterial isolates from S. lacustris collected in Lake Pichlinger See (Upper Austria) by the 16S rRNA gene sequencing, with the purpose of testing their biotechnological potentialities (see the following section). The phylogenetic analysis revealed that isolates were representatives of Gram-negative and Gram-positive bacteria within 28 and 13 genera, respectively. The former were mostly affiliated with Alphaproteobacteria (mainly in the genera Rhizobium and Brevundimonas), followed by Gammaproteobacteria (mainly affiliated with Pseudomonas spp.) and, at a lesser extent, by few Bacteroidetes and Verrucomicrobiota. Gram-positive isolates were mainly affiliated with Actinobacteria, including the genera Microbacterium, Frigoribacterium, Streptomyces, and Micrococcus. Bacillus and Exiguobacterium members were among Firmicutes isolates.
3.1.5. Tubella variabilis
By using five different culture media, Laport et al. [21] isolated 104 bacterial strains from T. variabilis (Bonetto and Ezcurra de Drago, 1973) (and an additional 59 isolates from freshwater) collected in an artificial channel receiving water from the da Prata River (Brazil). By the 16S rRNA gene sequencing, the strains were resolved in a total of 23 genera, with 12 of them that were isolated exclusively from the sponges. Most sponge-associated bacterial isolates were affiliated with Proteobacteria (namely Alpha-, Beta-, Delta-, Gammaproteobacteria classes). Gammaproteobacteria predominated in T. variabilis and were mainly represented by the family Enterobacteriaceae, with the genera Klebsiella and Enterobacter. The sole alphaproteobacterial isolate, belonging to the genus Methylobacterium, was isolated from a sponge sample. Among Firmicutes, which was the second most frequently isolated bacterial phylum, Fictibacillus and Bacillus were the predominant genera, with Bacillus that was not isolated from freshwater samples. Actinobacteria were also isolated only from sponges and were represented by a single isolate of the genera Microbacterium, Micrococcus, Rhodococcus, and Streptomyces. Finally, a unique Bacteroidetes isolate from T. variabilis was affiliated to the genus Chryseobacterium.
3.2. Biotechnological Relevant Bacteria from Freshwater Sponges
Recently, Clark et al. [42], combining MALDI-TOF mass spectrometry and the bioinformatics pipeline IDBac, analyzed both diversity and specialized molecule production of 692 bacterial strains isolated from Eunapius fragilis. Results highlighted the high biotechnological potential of bacteria isolates of freshwater sponges-associated bacteria. Notably, the specialized molecule production profiles varied at the bacterial species level and below, suggesting that in the search for novel bioactive compounds bacterial taxa should be analyzed on a case-by-case basis. To date, bacterial isolates from six freshwater sponge species have been targeted for their biotechnological potential as producers of molecules with antibiotic activities and enzymes (Table 3). Additional four sponge species have been targeted for the search of polyketide synthase (PKS) encoding genes within the whole microbial community (see below).
3.2.1. Potential for the Production of Bioactive Compounds by Bacterial Isolates
According to Keller-Costa et al., [25] freshwater sponges constitute a reservoir of Pseudomonas spp. (among Gammaproteobacteria) of biotechnological value due to their antimicrobial activity. For instance, novel polyketide synthase (PKS) encoding genes (codifying for a number of bioactive compounds) were reported for a Pseudomonas fluorescence isolate (namely, strain 28Bb08) from B. bacillifera [43]. The low homology level between the obtained amino acid gene sequences and the sequences of known biologically active compound synthases suggested that strain 28Bb08 probably produced a far undescribed bioactive compound. Later, Keller-Costa et al. [25] addressed the diversity and in vitro antimicrobial activities of Pseudomonas spp. isolated from E. fluviatilis, demonstrating their antibacterial, antiprotozoan, and antioomycetal activities. Some strains were also able to inhibit the growth of basidiomycetal and ascomycetal pests. This finding suggests that E. fluviatilis is a promising source of underexplored Pseudomonas strains. Their diversified bioactivity spectrum might characterize sponge-associated pseudomonads, with relevance in their functionality within the holobiont.
Axenov-Gribanov et al. [44] isolated and characterized actinobacterial strains from dominant benthic organisms’ communities of Lake Baikal, including B. bacillifera. Two isolates, namely Streptomyces sp. IB2014/01-2 and Pseudonocardia sp. IB2014/02-2, were screened for antibiotic activities against Bacillus subtilis, Staphylococcus carnosus, Pseudomonas putida, E. coli, and Saccharomyces cerevisiae. Both strains were able to inhibit the growth of S. cerevisiae and E. coli. C. albicans, and S. carnosus were inhibited by Streptomyces sp. IB2014/01-2 and Pseudonocardia sp. IB2014/02-2, respectively. The inhibitory activity varied based on the culture medium used for growth.
The 20.2% (21 out of 104) of bacterial isolates from T. variabilis showed inhibitory activity against Staphylococcus aureus [21]. Active bacteria were affiliated with the genera Aquitalea and Chromobacterium (both within Betaproteobacteria), Dickeya (within Gammaproteobacteria), and Klebsiella (within Enterobacteriaceae).
Graffius et al. [23] selected 33 isolates (representing 31 bacterial genera) from S. lacustris collected in Lake Pichlinger See (Upper Austria) for genome sequencing. Overall, results revealed the occurrence of 306 secondary metabolite biosynthesis gene clusters (BGCs). According to the authors, although using different culture media for bacterial isolation, the cultivable fraction did not reflect the whole bacterial community (as it was determined by metagenome and 16S rRNA gene amplicon sequencing analyses; see the section above for S. lacustris), suggesting that isolates may be either low-abundance representatives of the S. lacustris associated bacterial community or transient bacteria. Two Streptomyces isolates (namely strains SL203 and SL294; Actinobacteria) showed the highest secondary metabolite biosynthesis potential, as they harbored 28 and 23 BGCs, respectively. A more in-depth analysis of these two strains performed by the combination of genome mining and methanolic extract characterization revealed that they could be able to produce molecules with antibiotic activities. In the same study, among Actinobacteria, the genome Gordonia sp. SL306 harbored the highest number of nonribosomal peptides (NRPs) related to BGCs. However, any retrieved BGC was linked to known secondary metabolites [23]. Finally, Bacillus sp. SL112 (among Firmicutes) hosted 14 BGCs in its genomes. Eight BGCs showed high similarities with already known BGCs retrieved in other Bacillus isolates and linked to the synthesis of some polyketides, nonribosomally synthesized peptides (such as amylocyclicin, bacillibactin, bacillaene, bacilysin, difficidin, fengycin, macrolactin, surfactin) with antibacterial, antifungal, and cytotoxic activities. The authors suggested that Bacillus sp. SL112 might be among those bacterial symbionts that are involved in the protection of their sponge host from predators and infections [23].
Among fourteen bacterial strains isolated from the freshwater sponge Metania reticulata (Bowerbank, 1863) from the Negro River (Brazil), Rozas et al. [27] individuated two bacterial strains (namely, MERETb.761 and MERETb.762) showing antimicrobial activity. Both bacterial strains inhibited the fungus Aspergillus sp., whereas MERETb.762 also inhibited Staphylococcus aureus. Two fractions of the MERETb.762 extract inhibited the degranulation of rat basophilic leukemia (RBL–2H3) cells and corresponded chemically to nitroaromatic compounds. The production of immunosuppressants is commonly considered a bacterial strategy to avoid expulsion from their hosts [45]. Rozas et al. [27] supposed that MERETb.762 might utilize such a strategy to maintain its relationship with the host. The authors also suggested that performing immunosuppressive assays on isolated compounds could enhance the discovery of novel bioactive molecules.
3.2.2. Biosynthesis of Enzymes by Bacterial Isolates
Different enzyme activities were reported for unidentified bacterial isolates from the Lake Baikal sponges L. baicalensis and B. bacillifera [14]. Collagenase activity was in the range of 50–100% and 82–100% of bacteria isolated from L. baicalensis and B. bacillifera, respectively, followed by phosphatase (up to 90 and 64% of bacteria isolated from L. baicalensis and B. bacillifera, respectively) and phospholipase activities (e.g., 67% of L. baicalensis isolates). Caseinase activity was shown by 18 (B. bacillifera isolates) to 50% (L. baicalensis isolates) of the total tested strains. Lipase activity was detected in only 10–20% of bacterial strains isolates from both sponge species. According to the authors, based on these findings, sponge-associated bacteria, cleaving several organic compounds and thus supplying additional nutrients to their host, provide the sponge with food.
3.2.3. Polyketide Synthase Encoding Genes within the Associated Bacterial Communities
Polyketide synthases (PKS) are multifunctional enzymes responsible for the synthesis of low molecular weight bioactive metabolites. PKS sequences correspond to gene clusters in the microbial genomes. Therefore, the ability of microbial communities to produce bioactive molecules can be investigated by sequencing these genes. The amplification and subsequent sequencing of ketosynthase (KS) domain fragments of the PKS genes were used in the study of microbiomes of sponges L. baicalensis [46], S. papyracea [47], Rezinkovia echinata Efremova, 2004 [48], and B. fungiformis (Makuschok, 1927) [49]. In the first study, 15 PKS gene fragments, differing from each other by 35–65% by aminoacid sequences, were identified in the L. baicalensis microbial community [46] and related to Alpha-, Beta-, and Deltaproteobacteria, Verrucomicrobia, and Cyanobacteria. Some sequences were related to the genes involved in the biosynthesis of curacin A, stigmatellin, nostophycin, and cryptophycins. Interestingly, sequences showed a 50–82% with already known sequences. Later, the same authors identified 18 PKS gene fragments in the S. papyracea community metagenome [47]. The closest homologs belonged to the bacterial phyla Cyanobacteria, Proteobacteria (including Betaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria), and Acidobacteria. The PKS gene spectrum significantly differed between S. papyracea and L. baicalensis [46]. Kaluzhnaya and Itskovich [48] identified a higher number (i.e., 36) of unique sequences of the PKS-gene fragments in the microbiome of R. echinata. Sequences were 57.3–99.6% identical to those of various taxonomic groups of bacteria: Cyanobacteria (Scytonema sp, Crocosphaera watsonii, Cyanobium usitatum, Anabaena sp., Nostoc sp., Nostoc punctiforme, Trichormus variabilis, Synechococcus sp.), Verrucomicrobia (Opitutus terrae, Pedosphaera parvula), Betaproteobacteria (Piscinibacter aquaticus), Alphaproteobacteria (Bradyrhizobium sp.), Gammaproteobacteria (Alteromonadaceae family), Chloroflexi (Anaerolineae family), Firmicutes (Paenibacillus polymyxa) and Acidobacteria. Notably, some of the discovered PKS genes were previously retrieved in the microbiomes of the sponge species L. baicalensis [46] and S. papyracea [47]. Finally, recently Kaluzhnaya and Itskovich [49] found that the B. fungiformis-associated microbial community included microorganisms (including the genera Gemmatimonas, Rubrivivax, and Hydrogenophaga) that could be potential producers of biologically active substances. The authors retrieved eight unique sequences, most of which were highly identical (97–99%) to the genes of PKS of prokaryotes from “core” communities (phyla Cyanobacteria, Proteobacteria, Planctomycetes, Latescibacteria, and Gemmatimonadates) associated with other Baikal sponge species (reported above), collected in different years and different parts of the lake [46,47,48].
Overall, these findings suggested the co-evolution of the host sponge and symbiotic microflora in the lake. On the other hand, several phylogenetically close PKS sequences were representative of Baikal planktonic microorganisms, indicating a close interaction between the symbiotic microbiome and the community of the environment surrounding the sponge host. Finally, the PKS sequences retrieved in Baikal sponges generally showed a low similarity with already known genes codifying for biologically active compounds, suggesting that freshwater sponge-associated bacteria might produce novel metabolites of a polyketide nature.
Figure 3 depicts the different freshwater sponge species investigated in the field of microbial ecology and shows the geographical dislocation of treated sponges.
4. Conclusions
Freshwater sponges are distributed globally at all latitudes and include more than 240 described species. They colonize continental habitats, spanning from the Arctic to tropical rain forests, that embrace both lotic and lentic systems. However, unlike their marine counterparts, understanding the roles played by sponge microbiomes in freshwater ecosystems is still in its infancy (e.g., their involvement in nutrient cycling and ecosystem functioning) and has yet to be clarified to disentangle the ecological and evolutionary significance of these unique and largely under-investigated microbial communities.
Overall, available data suggest that freshwater sponge symbionts may be able to utilize sponge-derived compounds and provide nutrients to the sponge host. Moreover, studies addressing the cultivable bacterial symbionts have highlighted their biotechnological potentialities as producers of bioactive compounds, suggesting their involvement in promoting sponge health. Both culture-dependent and culture-independent (only recently including next-generation sequencing) methods have been applied to describe the freshwater sponge-associated bacterial communities, which result generally predominated by Proteobacteria, Bacteroidetes, and Actinobacteria, with differences encountered with respect to marine sponges. However, fragmentary data are available on the composition of the prokaryotic communities associated with only a few different sponge species, which inhabit distant locations worldwide. Moreover, very diversified methodological approaches have been used, thus making poorly comparable the available data and arduous an exhaustive and solid assessment of the topic. Therefore, testing associated microbiome for biogeographic variation in the freshwater sponges could be useful to assess if sponge species-microbe relationships are conserved in freshwater systems as in the marine environment, and individuate relevant environmental factors, along with anthropogenic stressors (e.g., pollution), involved in the acquisition and structuring of their microbiomes. Moreover, most studies describing the sponge-associated prokaryotic communities (targeting quite exclusively the bacterial fraction) lack also considering the natural environment (i.e., water, sediment, and biofilm) surrounding the analyzed sponge individuals. This approach should be more extensively applied in the future to better discriminate between transient and truly associated bacteria.
Given the aforementioned, freshwater sponges may represent neglected hotspots of complex bacterial biodiversity with functional and metabolic features, as well as potential in the production of promising biotechnologically relevant bioactive compounds, that remain yet to be discovered.
Author Contributions
Conceptualization, A.L.G.; methodology, A.L.G. and C.R.; software, C.R.; resources, A.L.G. and C.R.; data curation, A.L.G. and C.R.; writing—original draft preparation, A.L.G. and C.R.; writing—review and editing, A.L.G. and C.R.; project administration, A.L.G.; funding acquisition, A.L.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants from the INTERACT Transnational Access EU Program (funded by H2020) within the projects “BIP: Benthic filter-feeding Invertebrates from the Arctic as accumulators of Pollutants and tolerant bacterial communities” (grant agreement n. 730938) and “CIRCE: SearChIng for emeRging Contaminants in Sub-Arctic rivErs” (grant agreement n. 871120).
Data Availability Statement
Data are available upon request.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial and financial relationships that could be construed as a potential conflict of interest.
References
- Gernert, C.; Glöckner, F.O.; Krohne, G.; Hentschel, U. Microbial diversity of the freshwater sponge Spongilla lacustris. Microb. Ecol. 2005, 50, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Sugden, S.; Holert, J.; Cardenas, E.; Mohn, W.W.; Stein, L.Y. Microbiome of the freshwater sponge Ephydatia muelleri shares compositional and functional similarities with those of marine sponges. ISME J. 2022, 16, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.J. The functional roles of marine sponges. Estuar. Coast. Shelf Sci. 2008, 79, 341–353. [Google Scholar] [CrossRef]
- de Goeij, J.M.; van Oevelen, D.; Vermeij, M.J.; Osinga, R.; Middelburg, J.J.; de Goeij, A.F.; Admiraal, W. Surviving in a marine desert: The sponge loop retains resources within coral reefs. Science 2013, 342, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Folkers, M.; Rombouts, T. Sponges revealed: A synthesis of their overlooked ecological functions within aquatic ecosystems. In YOUMARES 9—The Oceans: Our Research, Our Future; Jungblut, S., Liebich, V., Bode-Dalby, M., Eds.; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Taylor, M.W.; Radax, R.; Steger, D.; Wagner, M. Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Moitinho-Silva, L.; Lurgi, M.; Björk, J.R.; Easson, C.; Astudillo-García, C.; Olson, J.B.; Erwin, P.M.; López-Legentil, S.; Luter, H.; et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 2016, 7, 11870. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, U.; Fieseler, L.; Wehrl, M.; Gernert, C.; Steinert, M.; Hacker, J.; Horn, M. Microbial diversity of marine sponges. In Marine Molecular Biotechnology; Mueller, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 59–88. [Google Scholar]
- Engelberts, J.P.; Robbins, S.J.; de Goeij, J.M.; Aranda, M.; Bell, S.C.; Webster, N.S. Characterization of a sponge microbiome using an integrative genome-centric approach. ISME J. 2020, 14, 1100–1110. [Google Scholar] [CrossRef]
- Webster, N.S.; Thomas, T. The sponge hologenome. MBio 2016, 7, e00135-16. [Google Scholar] [CrossRef]
- Manconi, R.; Pronzato, R. Global diversity of sponges (Porifera: Spongillina) in freshwater. Hydrobiologia 2008, 595, 27–33. [Google Scholar] [CrossRef]
- Erpenbeck, D.; Galitz, A.; Wörheide, G.; Albrecht, C.; Pronzato, R.; Manconi, R. Having the balls to colonize—The Ephydatia fluviatilis group and the origin of (ancient) Lake “endemic” sponge lineages. J. Gt. Lakes Res. 2020, 46, 1140–1145. [Google Scholar] [CrossRef]
- Wilkinson, C.R. Nutrient translocation from green algal symbionts to the freshwater sponge Ephydatia fluviatilis. Hydrobiologia 1980, 75, 241–250. [Google Scholar] [CrossRef]
- Parfenova, V.; Terkina, I.; Kostornova, T.Y.; Nikulina, I.G.; Chernykh, V.I.; Maksimova, E.A. Microbial community of freshwater sponges in Lake Baikal. Biol. Bull. 2008, 35, 374–379. [Google Scholar] [CrossRef]
- Kaluzhnaya, O.; Krivich, A.; Itskovich, V. Diversity of 16S rRNA genes in metagenomic community of the freshwater sponge Lubomirskia baicalensis. Russ. J. Genet. 2012, 48, 855–858. [Google Scholar] [CrossRef]
- Belikov, S.; Belkova, N.; Butina, T.; Chernogor, L.; Kley, A.M.; Nalian, A.; Rorex, C.; Khanaev, I.; Maikova, O.; Feranchuk, S. Diversity and shifts of the bacterial community associated with Baikal sponge mass mortalities. PLoS ONE 2019, 14, e0213926. [Google Scholar] [CrossRef] [PubMed]
- Chernogor, L.; Klimenko, E.; Khanaev, I.; Belikov, S. Microbiome analysis of healthy and diseased sponges Lubomirskia baicalensis by using cell cultures of primmorphs. Peer J. 2020, 8, e9080. [Google Scholar] [CrossRef] [PubMed]
- Petrushin, I.; Belikov, S.; Chernogor, L. Cooperative interaction of Janthinobacterium sp. Slb01 and Flavobacterium sp. slb02 in the diseased sponge Lubomirskia baicalensis. Int. J. Mol. Sci. 2020, 21, 8128. [Google Scholar] [CrossRef] [PubMed]
- Kulakova, N.V.; Sakirko, M.V.; Adelshin, R.V.; Khanaev, I.V.; Nebesnykh, I.A.; Pérez, T. Brown rot syndrome and changes in the bacterial сommunity of the Baikal sponge Lubomirskia baicalensis. Microb. Ecol. 2018, 75, 1024–1034. [Google Scholar] [CrossRef]
- Gaikwad, S.; Shouche, Y.S.; Gade, W.N. Microbial community structure of two freshwater sponges using Illumina MiSeq sequencing revealed high microbial diversity. AMB Express 2016, 6, 40. [Google Scholar] [CrossRef]
- Laport, M.S.; Pinheiro, U.; da Costa Rachid, C.T.C. Freshwater sponge Tubella variabilis presents richer microbiota than marine sponge species. Front. Microbiol. 2019, 10, 2799. [Google Scholar] [CrossRef]
- Kenny, N.J.; Francis, W.R.; Rivera-Vicéns, R.E.; Juravel, K.; de Mendoza, A.; Díez-Vives, C.; Lister, R.; Bezares-Calderón, L.A.; Grombacher, L.; Roller, M.; et al. Tracing animal genomic evolution with the chromosomal-level assembly of the freshwater sponge Ephydatia muelleri. Nat. Commun. 2020, 11, 3676. [Google Scholar] [CrossRef]
- Graffius, S.; Garzón, J.F.G.; Zehl, M.; Pjevac, P.; Kirkegaard, R.; Flieder, M.; Loy, A.; Rattei, T.; Ostrovsky, A.; Zotchev, S.B. Secondary metabolite production potential in a microbiome of the freshwater sponge Spongilla lacustris. Microbiol. Spectr. 2023, 11, e0435322. [Google Scholar] [CrossRef] [PubMed]
- Wiens, M.; Korzhev, M.; Perovic-Ottstadt, S.; Luthringer, B.; Brandt, D.; Klein, S.; Muller, W.E.G. Toll-like receptors are part of the innate immune defense system of sponges (Demospongiae: Porifera). Mol. Biol. Evol. 2007, 24, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Keller-Costa, T.; Jousset, A.; Van Overbeek, L.; Van Elsas, J.D.; Costa, R. The freshwater sponge Ephydatia fluviatilis harbours diverse Pseudomonas species (Gammaproteobacteria, Pseudomonadales) with broad-spectrum antimicrobial activity. PLoS ONE 2014, 9, e88429. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Rezanka, T.; Srebnik, M. Lipid compounds of freshwater sponges: Family Spongillidae class Demospongiae. Chem. Phys. Lip. 2003, 123, 117–155. [Google Scholar] [CrossRef] [PubMed]
- Rozas, E.E.; Mendes, M.A.; Nascimento, C.A.O.; Rodrigues, J.C.V.; Albano, R.M.; Custódio, M.R. Reduction of RBL–2H3 cells degranulation by nitroaromatic compounds from a Bacillus strain associated to the Amazonian sponge Metania reticulata. J. Mar. Biol. Assoc. U. K. 2016, 96, 567–572. [Google Scholar] [CrossRef]
- Costa, R.; Keller-Costa, T.; Gomes, N.C.M.; da Rocha, U.N.; van Overbeek, L.; van Elsas, J.D. Evidence for selective bacterial community structuring in the freshwater sponge Ephydatia fluviatilis. Microb. Ecol. 2013, 65, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Kaluzhnaya, O.V.; Itskovich, V.B.; McCormack, G.P. Phylogenetic diversity of bacteria associated with the endemic freshwater sponge Lubomirskia baicalensis. World J. Microbiol. Biotechnol. 2011, 27, 1955–1959. [Google Scholar] [CrossRef]
- Gladkikh, A.S.; Kalyuzhnaya, O.V.; Belykh, O.I.; Ahn, T.S.; Parfenova, V.V. Analysis of bacterial communities of two Lake Baikal endemic sponge species. Mikrobiologiia 2014, 83, 787–797. [Google Scholar] [CrossRef]
- Jung, D.; Seo, E.-Y.; Epstein, S.S.; Joung, Y.; Han, J.; Parfenova, V.V.; Belykh, O.I.; Gladkikh, A.S.; Ahn, T.S. Application of a new cultivation technology, I-tip, for studying microbial diversity in freshwater sponges of Lake Baikal, Russia. FEMS Microbiol. Ecol. 2014, 90, 417–423. [Google Scholar] [CrossRef]
- Seo, E.-Y.; Jung, D.; Belykh, O.I.; Bukshuk, N.A.; Parfenova, V.V.; Joung, Y.; Kim, I.C.; Yim, J.H.; Ahn, T.-S. Comparison of bacterial diversity and species composition in three endemic Baikalian sponges. Ann. Limnol.—Int. J. Lim. 2016, 52, 27–32. [Google Scholar] [CrossRef]
- Kenny, N.J.; Please, B.; Riesgo, A.; Itskovich, V.B. Symbiosis, selection, and novelty: Freshwater adaptation in the unique sponges of Lake Baikal. Mol. Biol. Evol. 2019, 36, 2462–2480. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.S.; Hammel, J.U.; Haen, K.M.; Danka, E.S.; Cieniewicz, B.; Winters, I.P.; Posfai, D.; Wörheide, G.; Lavrov, D.V.; Knight, S.W.; et al. RNA interference in marine and freshwater sponges: Actin knockdown in Tethya wilhelma and Ephydatia muelleri by ingested dsRNA expressing bacteria. BMC Biotechnol. 2011, 11, 67. [Google Scholar] [CrossRef] [PubMed]
- Hustus, K.; Díez-Vives, C.; Mitsi, K.; Nutakki, J.; Kering, V.; Nguyen, I.T.; Spencer, M.G.; Leys, S.P.; Hill, M.S.; Riesgo, A. Algal symbionts of the freshwater sponge Ephydatia muelleri. Symbiosis 2023, 90, 259–273. [Google Scholar] [CrossRef]
- Hall, C.; Camilli, S.; Dwaah, H.; Kornegay, B.; Lacy, C.; Hill, M.S.; Hill, A.L. Freshwater sponge hosts and their green algae symbionts: A tractable model to understand intracellular symbiosis. Peer J. 2021, 9, e10654. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.; Belikov, S.I.; Kaluzhnaya, O.V.; Perović-Ottstadt, S.; Fattorusso, E.; Ushijima, H.; Krasko, A.; Schröder, H.C. Cold stress defense in the freshwater sponge Lubomirskia baicalensis. Role of okadaic acid produced by symbiotic dinoflagellates. FEBS J. 2007, 274, 23–36. [Google Scholar] [CrossRef]
- Frost, T.M.; Williamson, C.E. In situ determination of the effect of symbiotic algae on the growth of the freshwater sponge Spongilla lacustris. Ecology 1980, 61, 1361–1370. [Google Scholar] [CrossRef]
- Sand-Jensen, K.; Pedersen, M.F. Photosynthesis by symbiotic algae in the fresh-water sponge, Spongilla lacustris. Limnol. Oceanogr. 1994, 39, 551–561. [Google Scholar] [CrossRef]
- Nicacio, G.; Pinheiro, U. Biodiversity of freshwater sponges (porifera: Spongillina) from northeast Brazil: New species and notes on systematics. Zootaxa 2015, 3981, 220–240. [Google Scholar] [CrossRef]
- Kohn, T.; Wiegand, S.; Boedeker, C.; Rast, P.; Heuer, A.; Jetten, M.S.M.; Schüler, M.; Becker, S.; Rohde, C.; Müller, R.W.; et al. Planctopirus ephydatiae, a novel Planctomycete isolated from a freshwater sponge. Syst. Appl. Microbiol. 2020, 43, 126022. [Google Scholar] [CrossRef]
- Clark, C.M.; Hernandez, A.; Mullowney, M.W.; Fitz-Henley, J.; Li, E.; Romanowski, S.B.; Pronzato, R.; Manconi, R.; Sanchez, L.M.; Murphy, B.T. Relationship between bacterial phylotype and specialized metabolite production in the culturable microbiome of two freshwater sponges. ISME Commun. 2022, 2, 22. [Google Scholar] [CrossRef]
- Lipko, I.A.; Kalyuzhnaya, O.V.; Kravchenko, O.S.; Parfenova, V.V. Identification of polyketide synthase genes in genome of Pseudomonas fluorescens strain 28Bb-06 from freshwater sponge Baikalospongia bacillifera. Mol. Biol. 2012, 46, 609–611. [Google Scholar] [CrossRef]
- Axenov-Gribanov, D.; Rebets, Y.; Tokovenko, B.; Voytsekhovskaya, I.; Timofeyev, M.; Luzhetskyy, A. The isolation and characterization of actinobacteria from dominant benthic macroinvertebrates endemic to Lake Baikal. Folia Microbiol. 2016, 61, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, D.; Venditti, D.; Padula, M.; Valassina, M. Intracellular life. Crit. Rev. Microbiol. 1999, 25, 39–79. [Google Scholar] [CrossRef] [PubMed]
- Kaluzhnaya, O.V.; Kulakova, N.V.; Itskovich, V.B. Diversity of polyketide synthase (PKS) genes in metagenomic community of freshwater sponge Lubomirskia baicalensis. Mol. Biol. 2012, 46, 790–795. [Google Scholar] [CrossRef]
- Kaluzhnaya, O.V.; Itskovich, V.B. Distinctive features of the microbial diversity and the polyketide synthase genes spectrum in the community of the endemic Baikal sponge Swartschewskia papyracea. Russ. J. Genet. 2016, 52, 38–48. [Google Scholar] [CrossRef]
- Kaluzhnaya, O.V.; Itskovich, V.B. Diversity of potential producers of bioactive metabolites having polyketide nature in the Baikal sponge community of Rezinkovia echinate. Limnol. Freshw. Biol. 2020, 3, 423–428. [Google Scholar] [CrossRef]
- Kaluzhnaya, O.V.; Itskovich, V.B. Features of diversity of polyketide synthase genes in the community of freshwater sponge Baikalospongia fungiformis. Russ. J. Genet. 2022, 58, 336–346. [Google Scholar] [CrossRef]
Figure 1.
Number of studies targeting bacterial communities associated with freshwater sponges.
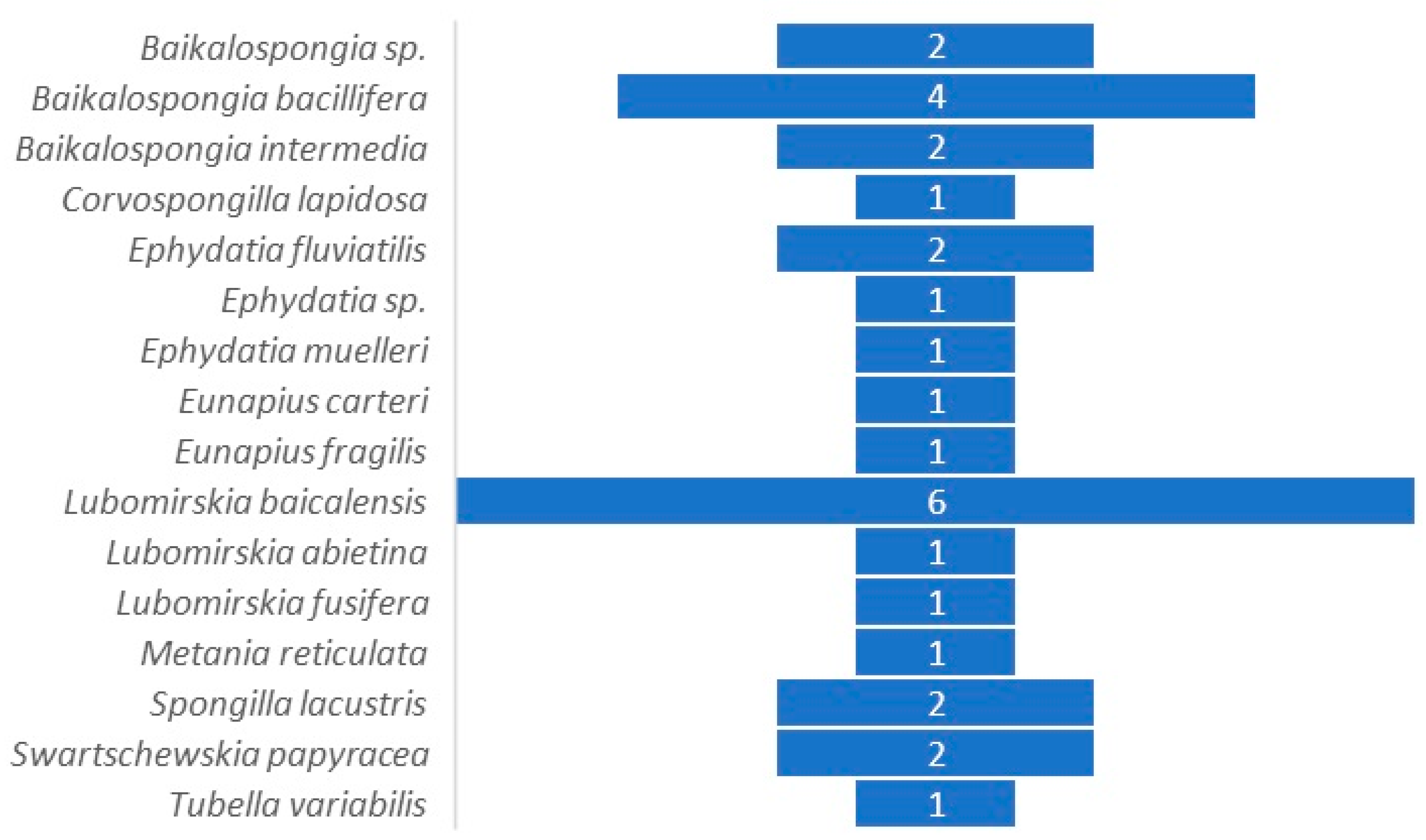
Figure 2.
Graphical representation of bacterial taxa detected in freshwater sponges from different sites. Presence/Absence matrix of bacterial taxa detected in different freshwater sponges (a); number of sponge specimens in which each taxon was detected (b); number of taxa detected in each sponge species/specimen (c): Baikalospongia intermedia [32], Baikalospongia sp. [30], Baikalospongia sp. [31], Corvospongilla lapidosa [20], Ephydatia fluvialis [25], Ephydatia muelleri [2], Eunapius carteri [20], Lubormirskia baicalensis sp. [30], Lubormirskia baicalensis [32], Lubormirskia baicalensis [31], Lubormirskia baicalensis [29], Spongilla lacustris [1], Spongilla lacustris [23], Swartschewskia papyracea [32], Tubella variabilis [21]; Shannon diversity index detected in bacterial communities associated with freshwater sponges (d): Baikalospongia sp. [30], Baikalospongia intermedia [32], Corvospongilla lapidosa [20], Ephydatia muelleri [2], Eunapius carteri [20], Lubormirskia baicalensis sp. [30], Lubormirskia baicalensis [32], Swartschewskia papyracea [32], Tubella variabilis [21]. Please note that Shannon Index is reported only when available from literature.
Figure 2.
Graphical representation of bacterial taxa detected in freshwater sponges from different sites. Presence/Absence matrix of bacterial taxa detected in different freshwater sponges (a); number of sponge specimens in which each taxon was detected (b); number of taxa detected in each sponge species/specimen (c): Baikalospongia intermedia [32], Baikalospongia sp. [30], Baikalospongia sp. [31], Corvospongilla lapidosa [20], Ephydatia fluvialis [25], Ephydatia muelleri [2], Eunapius carteri [20], Lubormirskia baicalensis sp. [30], Lubormirskia baicalensis [32], Lubormirskia baicalensis [31], Lubormirskia baicalensis [29], Spongilla lacustris [1], Spongilla lacustris [23], Swartschewskia papyracea [32], Tubella variabilis [21]; Shannon diversity index detected in bacterial communities associated with freshwater sponges (d): Baikalospongia sp. [30], Baikalospongia intermedia [32], Corvospongilla lapidosa [20], Ephydatia muelleri [2], Eunapius carteri [20], Lubormirskia baicalensis sp. [30], Lubormirskia baicalensis [32], Swartschewskia papyracea [32], Tubella variabilis [21]. Please note that Shannon Index is reported only when available from literature.

Figure 3.
Freshwater sponges investigated in the field of associated prokaryotic communities and their geographical dislocation.
Figure 3.
Freshwater sponges investigated in the field of associated prokaryotic communities and their geographical dislocation.
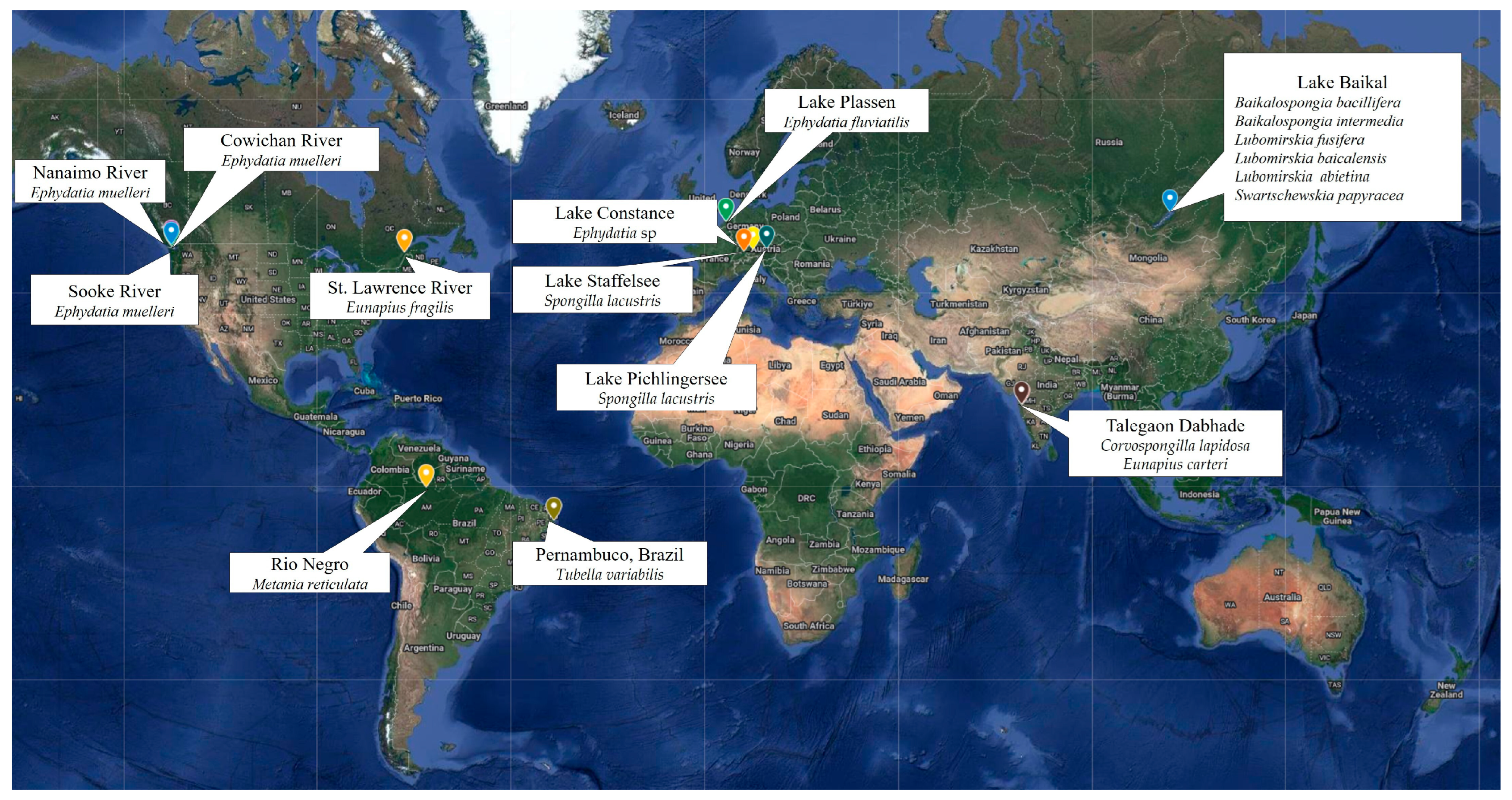
Table 2.
Studies investigating the cultivable fraction of the freshwater sponge-associated bacterial communities.
Table 2.
Studies investigating the cultivable fraction of the freshwater sponge-associated bacterial communities.
| Sponge Species | Freshwater Sampling Site | Reference |
| Baikalospongia bacillifera (Dybowski, 1880) | Lake Baikal (Russia) | [14] |
| Baikalospongia intermedia (Dybowsky, 1880) | Lake Baikal (Russia) | [14] |
| Ephydatia sp. | Lake Constance (Germany) | [41] |
| Eunapius fragilis (Leidy, 1851) | St. Lawrence River (North America) | [42] |
| Lubomirskia fusifera (Soukatschoff, 1895) | Lake Baikal (Russia) | [14] |
| Lubomirskia baicalensis (Pallas, 1776) | Lake Baikal (Russia) | [14] |
| Spongilla lacustris (Linnaeus, 1759) | Pichlinger See (Austria) | [23] |
| Swartschewskia papyracea (Dybowsky, 1880) | Lake Baikal (Russia) | [14] |
| Tubella variabilis (Bonetto and Ezcurra de Drago, 1973) | Artificial channel (Brazil) | [21] |
Table 3.
Bacterial isolates of biotechnological interest from freshwater sponges.
| Sponge Species | Freshwater Sampling Site | Bacterial Isolate(s) ID | Activity | Reference |
|---|---|---|---|---|
| B. bacillifera | Lake Baikal | Pseudomonas strain 28Bb08 | Detection of PKS genes | [43] |
| Lake Baikal | Unidentified | Enzyme activity | [14] | |
| Lake Baikal | Streptomyces sp. IB2014/01-2; Pseudonocardia sp. IB2014/02-2 | Antibiotic activities | [44] | |
| E. fluviatilis | Vinkeveense Plassen Lake (The Netherlands) | Pseudomonas spp. | Antibiotic activities | [25] |
| L. baicalensis | Lake Baikal | Unidentified | Enzyme activity | [14] |
| M. reticulata | Negro River (Brazil) | Bacillus strain MERETb.762 | Antibiotic activities | [27] |
| S. lacustris | Pichlinger See, Upper Austria | Bacillus sp. SL112 | Antibiotic activities | [23] |
| Pichlinger See, Upper Austria | Gordonia sp. SL306 | Antibiotic activities | [23] | |
| Pichlinger See, Upper Austria | Streptomyces spp. SL203 and SL294 | Antibiotic activities | [23] | |
| T. variabilis | Artificial channel (Brazil) | Several genera | Antibiotic activities | [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lo Giudice, A.; Rizzo, C. Freshwater Sponges as a Neglected Reservoir of Bacterial Biodiversity. Microorganisms 2024, 12, 25. https://doi.org/10.3390/microorganisms12010025
AMA Style
Lo Giudice A, Rizzo C. Freshwater Sponges as a Neglected Reservoir of Bacterial Biodiversity. Microorganisms. 2024; 12(1):25. https://doi.org/10.3390/microorganisms12010025
Chicago/Turabian StyleLo Giudice, Angelina, and Carmen Rizzo. 2024. "Freshwater Sponges as a Neglected Reservoir of Bacterial Biodiversity" Microorganisms 12, no. 1: 25. https://doi.org/10.3390/microorganisms12010025
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.