Proteomic Characterization of Armillaria mellea Reveals Oxidative Stress Response Mechanisms and Altered Secondary Metabolism Profiles

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture Conditions
2.2. Protein Extraction and 2-DE
2.3. Protein Extraction for In-Solution Digestion
2.4. LC-MS/MS Identification of A. mellea Proteins
2.5. A. mellea Metabolomics
2.6. Bioinformatic Tools
3. Results
3.1. 2-DE Interpretation
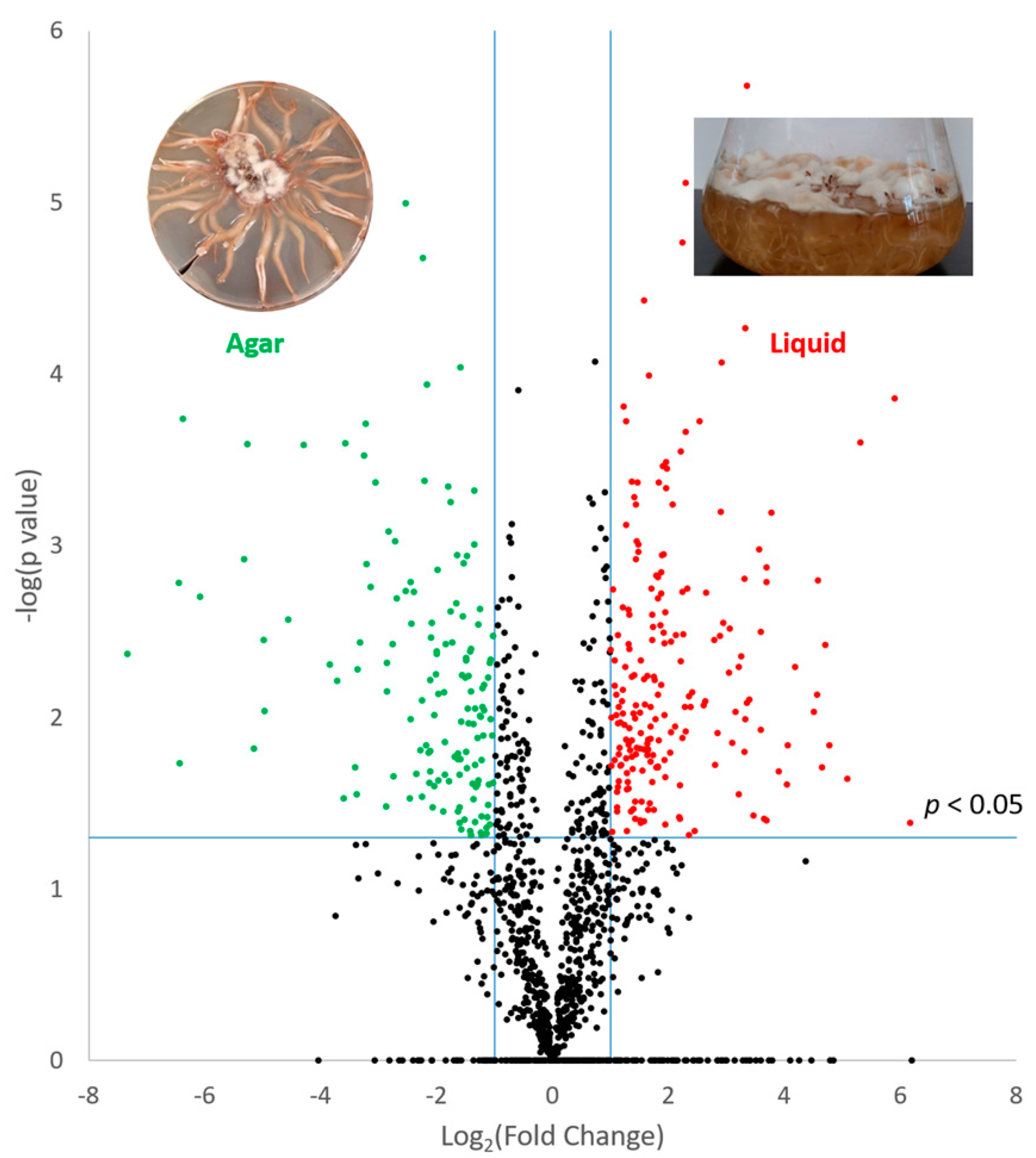
3.2. Effect of Culture Matrix on Protein Expression in A. mellea
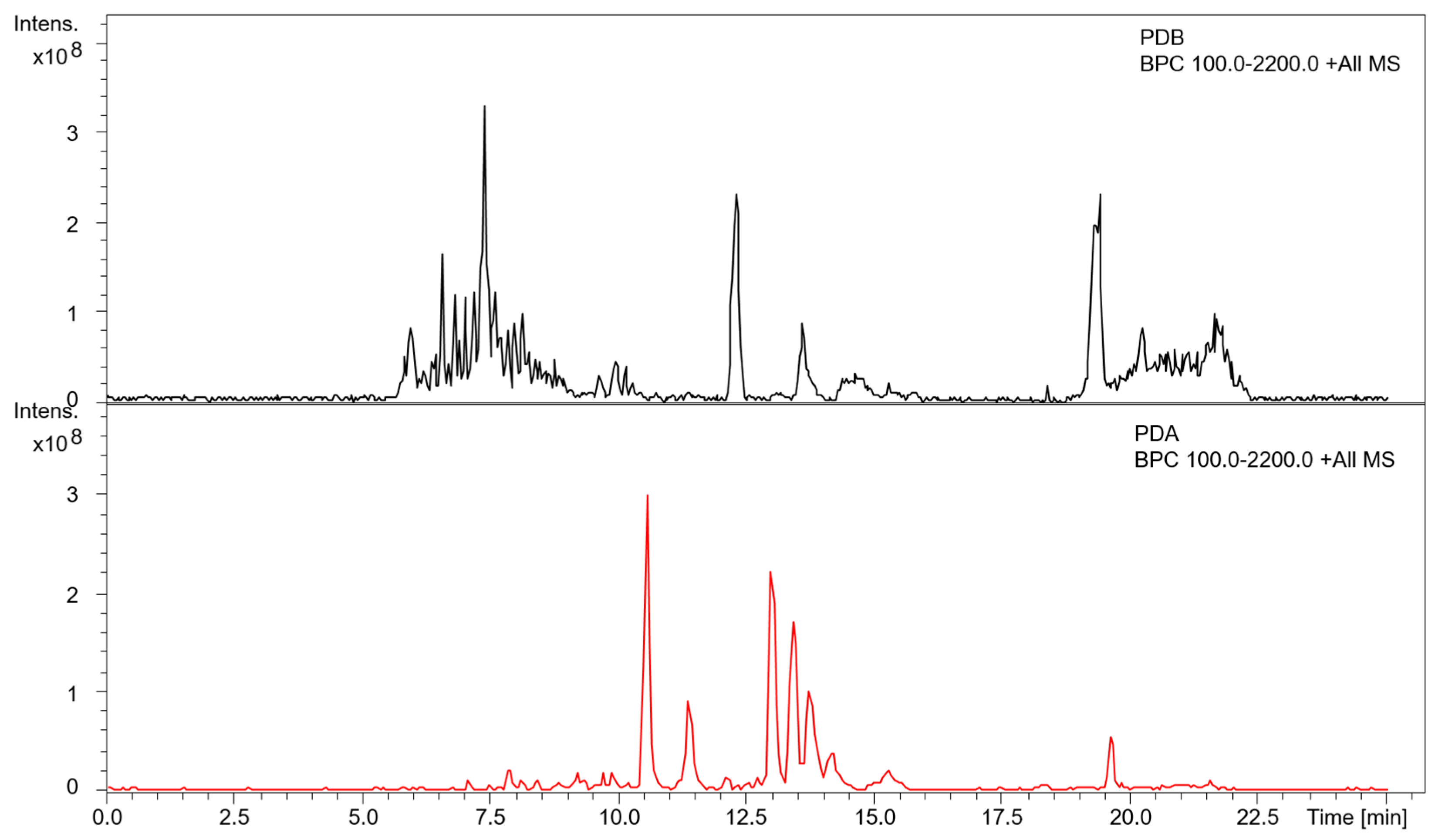
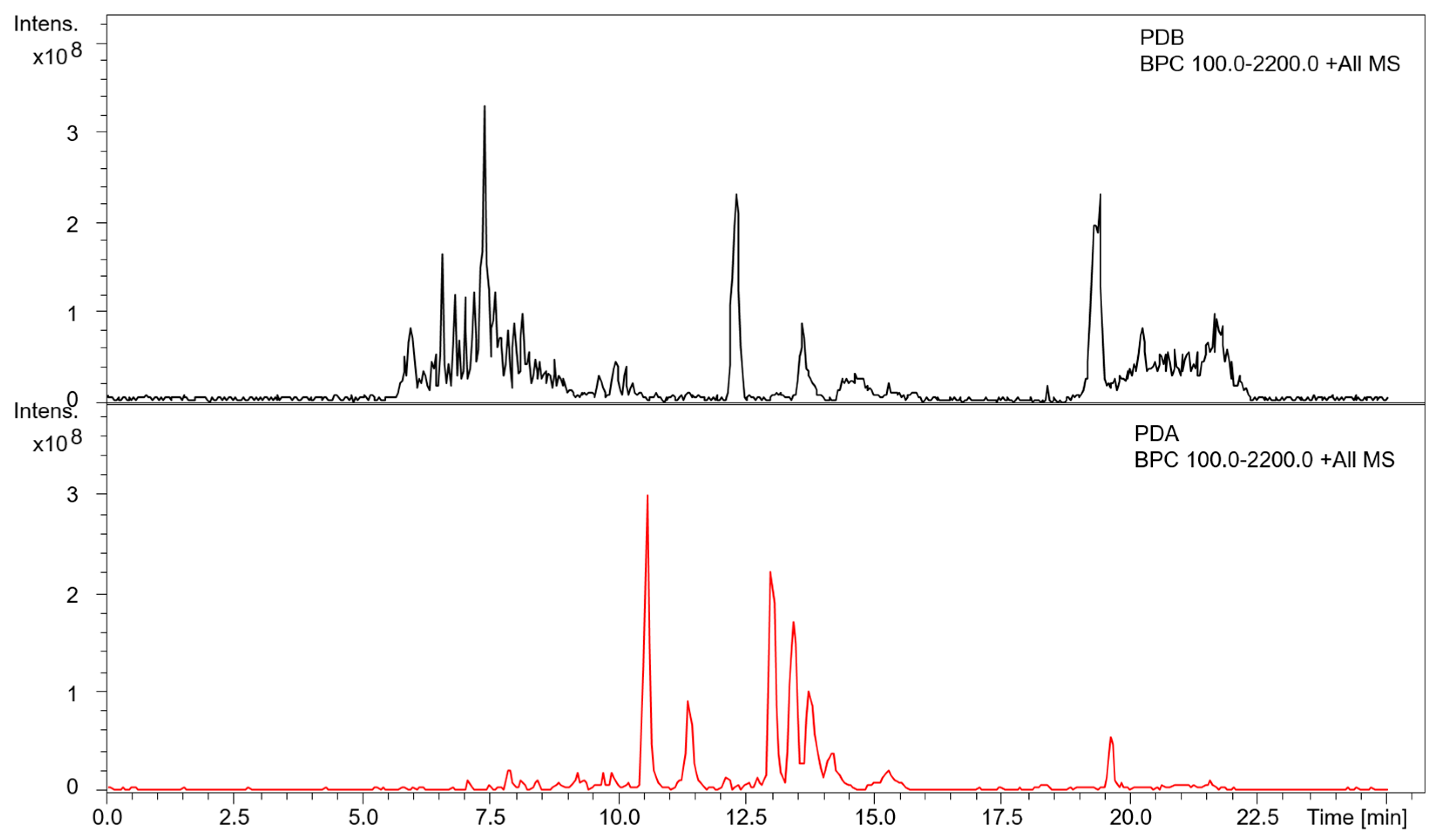
3.3. Metabolite Profiling of A. mellea in Different Culture Conditions
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, Q.; Harvey, L.M.; McNeil, B. Oxidative stress in industrial fungi. Crit. Rev. Biotechnol. 2009, 29, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.; Tudzynski, P. Reactive oxygen species in phytopathogenic fungi: signaling, development, and disease. Annu. Rev. Phytopathol. 2011, 49, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Zhaxybayeva, O.; Papke, R.T.; Doolittle, W.F. Actinorhodopsins: proteorhodopsin-like gene sequences found predominantly in non-marine environments. Environ. Microbiol. 2008, 10, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Syed, K.; Yadav, J.S. P450 monooxygenases (P450ome) of the model white rot fungus Phanerochaete chrysosporium. Crit. Rev. Microbiol. 2012, 38, 339–363. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Leonowicz, A.; Cho, N.S.; Luterek, J.; Wilkolazka, A.; Wojtas-Wasilewska, M.; Matuszewska, A.; Hofrichter, M.; Wesenberg, D.; Rogalski, J. Fungal laccase: properties and activity on lignin. J. Basic Microbiol. 2001, 41, 185–227. [Google Scholar] [CrossRef]
- Hammel, K.E.; Cullen, D. Role of fungal peroxidases in biological ligninolysis. Curr. Opin. Plant Biol. 2008, 11, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Lundell, T.K.; Makela, M.R.; Hilden, K. Lignin-modifying enzymes in filamentous basidiomycetes--ecological, functional and phylogenetic review. J. Basic Microbiol. 2010, 50, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Pocsi, I.; Miskei, M.; Karanyi, Z.; Emri, T.; Ayoubi, P.; Pusztahelyi, T.; Balla, G.; Prade, R.A. Comparison of gene expression signatures of diamide, H2O2 and menadione exposed Aspergillus nidulans cultures--linking genome-wide transcriptional changes to cellular physiology. BMC Genomics 2005, 6, 182. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Toledano, M.B.; Delaunay, A.; Biteau, B.; Spector, D.; Azevedo, D. Oxidative responses in yeast. In Yeast Stress Responses; Hohmann, S., Mager, W.H., Eds.; Springer: New York, NY, USA, 2003; pp. 241–303. [Google Scholar]
- Alfaro, M.; Oguiza, J.A.; Ramírez, L.; Pisabarro, A.G. Comparative analysis of secretomes in basidiomycete fungi. J. Proteomics 2014, 102, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Fernandez, R.; Jorrin-Novo, J.V. Contribution of proteomics to the study of plant pathogenic fungi. J. Proteome Res. 2012, 11, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Bianco, L.; Perrotta, G. Methodologies and perspectives of proteomics applied to filamentous fungi: From sample preparation to secretome analysis. Int. J. Mol. Sci. 2015, 16, 5803–5829. [Google Scholar] [CrossRef] [PubMed]
- Vasina, D.V.; Pavlov, A.R.; Koroleva, O.V. Extracellular proteins of Trametes hirsuta st. 072 induced by copper ions and a lignocellulose substrate. BMC Microbiol. 2016, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Doré, J.; Perraud, M.; Dieryckx, C.; Kohler, A.; Morin, E.; Henrissat, B.; Lindquist, E.; Zimmermann, S.D.; Girard, V.; Kuo, A.; et al. Comparative genomics, proteomics and transcriptomics give new insight into the exoproteome of the basidiomycete Hebeloma cylindrosporum and its involvement in ectomycorrhizal symbiosis. New Phytol. 2015, 208, 1169–1187. [Google Scholar] [CrossRef]
- Matsuzaki, F.; Shimizu, M.; Wariishi, H. Proteomic and metabolomic analyses of the white-rot fungus phanerochaete chrysosporium exposed to exogenous benzoic acid. J. Proteome Res. 2008, 7, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Imanaka, H.; Tanaka, S.; Feng, B.; Imamura, K.; Nakanishi, K. Cultivation characteristics and gene expression profiles of Aspergillus oryzae by membrane-surface liquid culture, shaking-flask culture, and agar-plate culture. J. Biosci. Bioeng. 2010, 109, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Keane, T.M.; Turner, D.J.; O’Keeffe, G.; Fitzpatrick, D.A.; Doyle, S. Genomic and Proteomic Dissection of the Ubiquitous Plant Pathogen, Armillaria mellea: Toward a New Infection Model System. J. Proteome Res. 2013, 12, 2552–2570. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2007, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Moloney, N.M.; Owens, R.A.; Meleady, P.; Henry, M.; Dolan, S.K.; Mulvihill, E.; Clynes, M.; Doyle, S. The iron-responsive microsomal proteome of Aspergillus fumigatus. J. Proteomics 2016, 136, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.A.; O’Keeffe, G.; Smith, E.B.; Dolan, S.K.; Hammel, S.; Sheridan, K.J.; Fitzpatrick, D.A.; Keane, T.M.; Jones, G.W.; Doyle, S. Interplay between Gliotoxin Resistance, Secretion and the Methyl/Methionine Cycle in Aspergillus fumigatus. Eukaryot. Cell 2015, 14, EC.00055-15. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Smedsgaard, J. Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. J. Chromatogr. A 1997, 760, 264–270. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. AntiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Wick, J.; Heine, D.; Lackner, G.; Misiek, M.; Tauber, J.; Jagusch, H.; Hertweck, C.; Hoffmeister, D. A fivefold parallelized biosynthetic process secures chlorination of Armillaria mellea (honey mushroom) toxins. Appl. Environ. Microbiol. 2016, 82, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Lackner, G.; Misiek, M.; Braesel, J.; Hoffmeister, D. Genome mining reveals the evolutionary origin and biosynthetic potential of basidiomycete polyketide synthases. Fungal Genet. Biol. 2012, 49, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Lackner, G.; Bohnert, M.; Wick, J.; Hoffmeister, D. Assembly of Melleolide Antibiotics Involves a Polyketide Synthase with Cross-Coupling Activity. Chem. Biol. 2013, 20, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Levine, R.L. Methionine in proteins defends against oxidative stress. FASEB J. 2009, 23, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Cowie, D.B.; Cohen, G.N.; Bolton, E.T.; De Robichon-Szulmajster, H. Amino acid analog incorporation into bacterial proteins. Biochim. Biophys. Acta 1959, 34, 39–46. [Google Scholar] [CrossRef]
- Suliman, H.S.; Appling, D.R.; Robertus, J.D. The Gene for Cobalamin-independent Methionine Synthase is Essential in Candida albicans: A Potential Antifungal Target. Arch. Biochem. Biophys. 2007, 467, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Kuuskeri, J.; Häkkinen, M.; Laine, P.; Smolander, O.-P.; Tamene, F.; Miettinen, S.; Nousiainen, P.; Kemell, M.; Auvinen, P.; Lundell, T. Time-scale dynamics of proteome and transcriptome of the white-rot fungus Phlebia radiata: growth on spruce wood and decay effect on lignocellulose. Biotechnol. Biofuels 2016, 9, 192. [Google Scholar] [CrossRef] [PubMed]
- Prado, R.S.; Bailão, A.M.; Silva, L.C.; de Oliveira, C.M.; Marques, M.F.; Silva, L.P.; Silveira-Lacerda, E.P.; Lima, A.P.; Soares, C.M.; Pereira, M. Proteomic profile response of Paracoccidioides lutzii to the antifungal argentilactone. Front. Microbiol. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hondorp, E.R.; Matthews, R.G. Oxidative stress inactivates cobalamin-independent methionine synthase (MetE) in Escherichia coli. PLoS Biol. 2004, 2, e336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leichert, L.I.; Jakob, U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004, 2, e333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hondorp, E.R.; Matthews, R.G. Oxidation of cysteine 645 of cobalamin-independent methionine synthase causes a methionine limitation in Escherichia coli. J. Bacteriol. 2009, 191, 3407–3410. [Google Scholar] [CrossRef] [PubMed]
- Rider, J.E.; Hacker, A.; Mackintosh, C.A.; Pegg, A.E.; Woster, P.M.; Casero, R.A.J. Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide. Amino Acids 2007, 33, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; McCann, P.P. Polyamine metabolism and function. Am. J. Physiol. 1982, 243, C212–C221. [Google Scholar] [PubMed]
- Hashimoto, T.; Tamaki, K.; Suzuki, K.; Yamada, Y. Molecular cloning of plant spermidine synthases. Plant Cell Physiol. 1998, 39, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Leon-Ramirez, C.G.; Valdes-Santiago, L.; Campos-Gongora, E.; Ortiz-Castellanos, L.; Arechiga-Carvajal, E.T.; Ruiz-Herrera, J. A molecular probe for Basidiomycota: the spermidine synthase-saccharopine dehydrogenase chimeric gene. FEMS Microbiol. Lett. 2010, 312, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Santiago, L.; Cervantes-Chavez, J.A.; Ruiz-Herrera, J. Ustilago maydis spermidine synthase is encoded by a chimeric gene, required for morphogenesis, and indispensable for survival in the host. FEMS Yeast Res. 2009, 9, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, S.; Kadoma, Y. Kinetic evaluation of polyamines as radical scavengers. Anticancer Res. 2005, 25, 965–969. [Google Scholar] [PubMed]
- Ha, H.C.; Yager, J.D.; Woster, P.A.; Casero, R.A.J. Structural specificity of polyamines and polyamine analogues in the protection of DNA from strand breaks induced by reactive oxygen species. Biochem. Biophys. Res. Commun. 1998, 244, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Aguilera, J.A.; Ward, J.F.; Fahey, R.C. Effect of polyamine-induced compaction and aggregation of DNA on the formation of radiation-induced strand breaks: quantitative models for cellular radiation damage. Radiat. Res. 1997, 148, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.K.; Chen, W.; Poy, G.; Cam, M.; Stiles, D.; Tabor, H. Microarray studies on the genes responsive to the addition of spermidine or spermine to a Saccharomyces cerevisiae spermidine synthase mutant. Yeast 2009, 26, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Lessing, F.; Kniemeyer, O.; Wozniok, I.; Loeffler, J.; Kurzai, O.; Haertl, A.; Brakhage, A.A. The Aspergillus fumigatus transcriptional regulator AfYap1 represents the major regulator for defense against reactive oxygen intermediates but is dispensable for pathogenicity in an intranasal mouse infection model. Eukaryot. Cell 2007, 6, 2290–2302. [Google Scholar] [CrossRef] [PubMed]
- Bohm, S.; Lamberti, G.; Fernandez-Saiz, V.; Stapf, C.; Buchberger, A. Cellular functions of Ufd2 and Ufd3 in proteasomal protein degradation depend on Cdc48 binding. Mol. Cell. Biol. 2011, 31, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Wilkinson, A.J. AAA+ superfamily ATPases: common structure--diverse function. Genes Cells 2001, 6, 575–597. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, C.; Rowicka, M.; Kudlicki, A.; Nowis, D.; McConnell, E.; Kujawa, M.; DeMartino, G.N. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 2006, 17, 4606–4618. [Google Scholar] [CrossRef] [PubMed]
- Dreveny, I.; Pye, V.E.; Beuron, F.; Briggs, L.C.; Isaacson, R.L.; Matthews, S.J.; McKeown, C.; Yuan, X.; Zhang, X.; Freemont, P.S. p97 and close encounters of every kind: a brief review. Biochem. Soc. Trans. 2004, 32, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Takata, T.; Kimura, Y.; Manno, A.; Murakami, K.; Koike, M.; Ohizumi, H.; Hori, S.; Kakizuka, A. ATPase activity of p97/valosin-containing protein is regulated by oxidative modification of the evolutionally conserved cysteine 522 residue in walker a motif. J. Biol. Chem. 2005, 280, 41332–41341. [Google Scholar] [CrossRef] [PubMed]
- Saric, T.; Graef, C.I.; Goldberg, A.L. Pathway for degradation of peptides generated by proteasomes: a key role for thimet oligopeptidase and other metallopeptidases. J. Biol. Chem. 2004, 279, 46723–46732. [Google Scholar] [CrossRef] [PubMed]
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim. Biophys. Acta - Mol. Cell Res. 2002, 1592, 63–77. [Google Scholar] [CrossRef]
- Goodwin, S.B.; M’barek, S.B.; Dhillon, B.; Wittenberg, A.H.J.; Crane, C.F.; Hane, J.K.; Foster, A.J.; Van der Lee, T.A.J.; Grimwood, J.; Aerts, A.; et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011, 7, e1002070. [Google Scholar] [CrossRef] [PubMed]
- Ralat, L.A.; Ren, M.; Schilling, A.B.; Tang, W.J. Protective role of Cys-178 against the inactivation and oligomerization of human insulin-degrading enzyme by oxidation and nitrosylation. J. Biol. Chem. 2009, 284, 34005–34018. [Google Scholar] [CrossRef] [PubMed]
- Shinall, H.; Song, E.S.; Hersh, L.B. Susceptibility of Amyloid Peptide Degrading Enzymes to Oxidative Damage: A Potential Alzheimer’ s Disease Spiral. Biochemistry 2005, 44, 15345–15350. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lapham, A.N.; Freedman, C.G.K.; Reed, T.L.; Schmidt, W.K. Yeast as a tractable genetic system for functional studies of the insulin-degrading enzyme. J. Biol. Chem. 2005, 280, 27481–27490. [Google Scholar] [CrossRef] [PubMed]
- Adames, N.; Blundell, K.; Ashby, M.N.; Boone, C. Role of yeast insulin-degrading enzyme homologs in propheromone processing and bud site selection. Science (80-) 1995, 270, 464–467. [Google Scholar] [CrossRef]
- Taskin, A.A.; Kücükköse, C.; Burger, N.; Mossmann, D.; Meisinger, C.; Vögtle, F.-N. The novel mitochondrial matrix protease Ste23 is required for efficient presequence degradation and processing. Mol. Biol. Cell 2017, 28, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Zhu, J.; Hu, X.; Pei, D.; Bell, C.E. Crystal structure of S-ribosylhomocysteinase (LuxS) in complex with a catalytic 2-ketone intermediate. Biochemistry 2005, 44, 3745–3753. [Google Scholar] [CrossRef] [PubMed]
- Parveen, N.; Cornell, K.A. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 2011, 79, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, B.A.; Weretilnyk, E.A. Sustaining S-adenosyl-l-methionine-dependent methyltransferase activity in plant cells. Physiol. Plant. 2001, 113, 435–442. [Google Scholar] [CrossRef]
- Owens, R.A.; Hammel, S.; Sheridan, K.J.; Jones, G.W.; Doyle, S. A Proteomic Approach to Investigating Gene Cluster Expression and Secondary Metabolite Functionality in Aspergillus fumigatus. PLoS One 2014, 9, e106942. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M.; Miethbauer, S.; Dahse, H.M.M.H.-M.; Ziemen, J.; Nett, M.; Hoffmeister, D. In vitro cytotoxicity of melleolide antibiotics: structural and mechanistic aspects. Bioorg. Med. Chem. Lett. 2011, 21, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M.; Nützmann, H.W.; Schroeckh, V.; Horn, F.; Dahse, H.M.; Brakhage, A.A.; Hoffmeister, D. Cytotoxic and antifungal activities of melleolide antibiotics follow dissimilar structure-activity relationships. Phytochemistry 2014, 105, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M.; Scherer, O.; Wiechmann, K.; König, S.; Dahse, H.M.; Hoffmeister, D.; Werz, O. Melleolides induce rapid cell death in human primary monocytes and cancer cells. Bioorganic Med. Chem. 2014, 22, 3856–3861. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, P.; Guo, C.; Palmer, J.M.; Sekonyela, R.; Wang, C.C.C. Prototype of an intertwined secondary- metabolite supercluster. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17065–17070. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.M.; Alberti, F.; Kilaru, S.; Collins, C.M.; de Mattos-Shipley, K.; Hartley, A.J.; Hayes, P.; Griffin, A.; Lazarus, C.M.; Cox, R.J.; et al. Identification and manipulation of the pleuromutilin gene cluster from Clitopilus passeckerianus for increased rapid antibiotic production. Sci. Rep. 2016, 6, 25202. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Funk, A.N.; Scherlach, K.; Horn, F.; Schroeckh, V.; Chankhamjon, P.; Westermann, M.; Roth, M.; Brakhage, A.A.; Hertweck, C.; et al. Differential expression of silent polyketide biosynthesis gene clusters in chemostat cultures of Aspergillus nidulans. J. Biotechnol. 2012, 160, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Heinig, U.; Grothe, T.; Stadler, M.; Jennewein, S.C.-P. Cloning and characterization of an Armillaria gallica cDNA encoding protoilludene synthase, which catalyzes the first committed step in the synthesis of antimicrobial melleolides. J. Biol. Chem. 2011, 286, 6871–6878. [Google Scholar] [CrossRef] [PubMed]
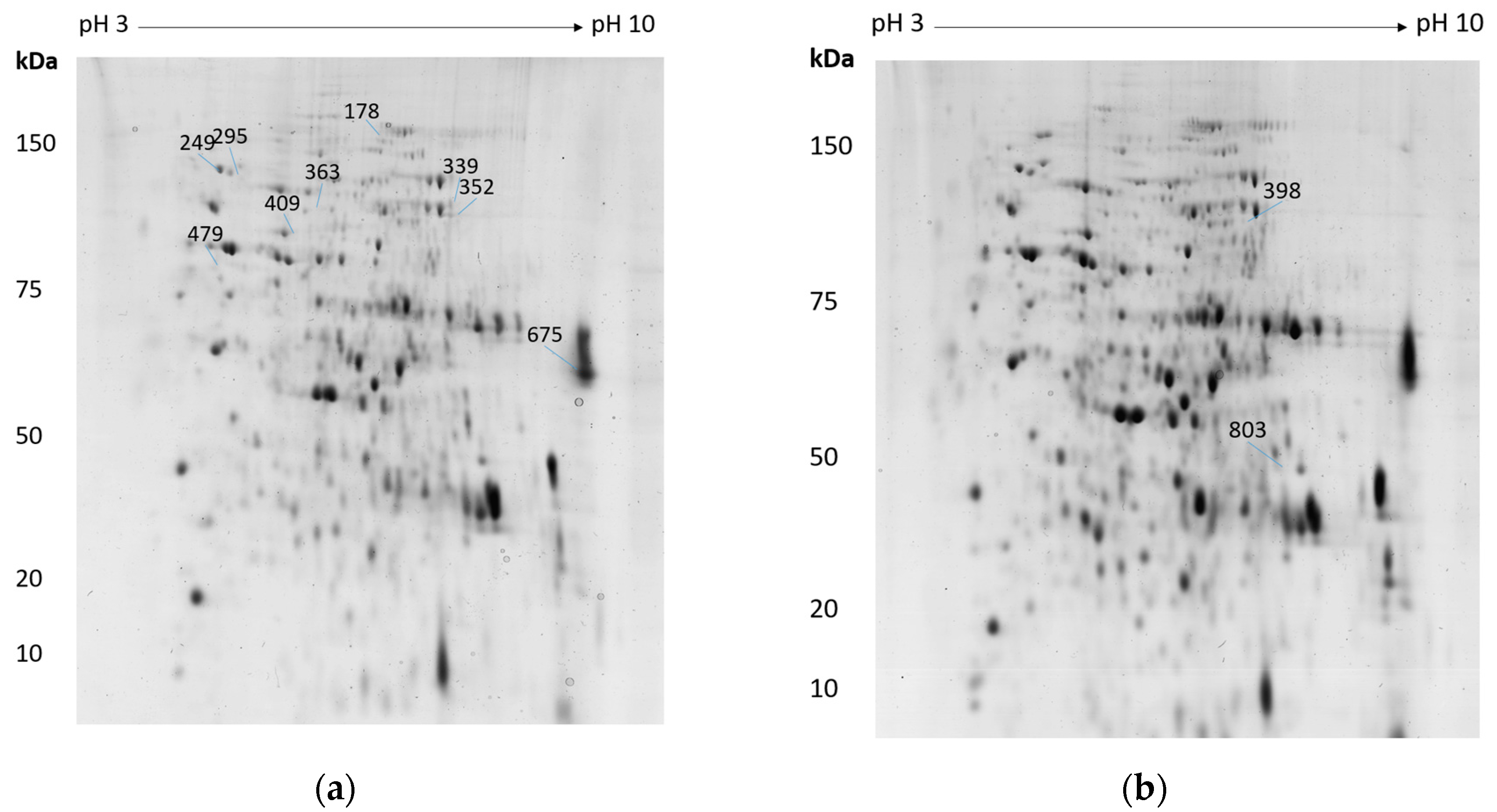


{kind=link}
{kind=link}
{kind=link}
| H2O2 | Menadione/FeCl3 | |||||
|---|---|---|---|---|---|---|
| Spot No. | a Accession No. | b BLAST Description | ANOVA (p) | c Fold Change | ANOVA (p) | c Fold Change |
| 339/398 | Am17277 | Cobalamin-independent methionine synthase | 0.032 | ↑ 2.7 | 0.028 | ↑ 2.6 |
| 352 | Am17277 | Cobalamin-independent methionine synthase | 0.005 | ↑ 1.8 | ||
| 409 | Am3212 | Zinc metallopeptidase found in the cytoplasm and intermembrane space of mitochondria | 0.008 | ↑ 1.8 | ||
| 363 | Am14050 | Saccharopine dehydrogenase | 0.036 | ↑ 1.7 | ||
| 249 | Am14558 | Valosin-containing protein | 0.017 | ↑ 1.6 | ||
| 295 | Am18454 | Heat shock protein | 0.032 | ↑ 1.6 | ||
| 803 | Am19877 | Glutamic oxaloacetic transaminase aat1 | 0.011 | ↑ 1.5 | ||
| 178 | Am16706 | A-pheromone processing metallopeptidase ste23 | 0.044 | ↓ 2.3 | ||
| 675 | Am19873 | Translation elongation factor 1a | 0.012 | ↓ 1.8 | ||
| 479 | Am7452 | Heat shock protein 90 | 0.034 | ↓ 1.5 | ||
| a Accession No. | b BLAST Description | p-Value | c Fold Change | Uniquely Detected | Unique Peptides | d Cluster No. | Putative Product |
|---|---|---|---|---|---|---|---|
| Increased in Agar | |||||||
| Am6587 | Fatty acid synthase | 0.002 | 5.8 | 111 | 1.1 | ||
| Am14527 | Polyketide synthase | ns | Agar | 3 | 1.4 | ||
| Am14528 | Polyketide synthase | ns | Agar | 2 | 1.4 | ||
| Am18600 | Aldo keto reductase | ns | Agar | 2 | 1.11 | ||
| Am19612 | Acetyl-synthetase | ns | Agar | 5 | 1.22 | ||
| Am15263 | Acetyl-synthetase | ns | Agar | 4 | 1.24 | ||
| Am315 | Polyketide synthase | ns | Agar | 7 | 1.25 | ||
| Am14855 | Cytochrome P450 | ns | Agar | 2 | 1.29 | Protoilludene/melleolides [28] | |
| Increased in Liquid | |||||||
| Am19046 | RNA-binding domain-containing | 0.016 | 2.7 | 11 | 1.3 | ||
| Am14843 | NAD(P)-binding | 0.015 | 2.2 | 10 | 1.28 | Orsellinic acid/melleolides [29,30] | |
| No significant Change | |||||||
| Am19045 | Glycosyltransferase family 20 | ns | 13 | 1.3 | |||
| Am14526 | Acetyl-synthetase | ns | 18 | 1.4 | |||
| Am12922 | Alpha aminoadipate reductase Lys1 | ns | 12 | 1.9 | |||
| Am18601 | Glycoside hydrolase family 7 | ns | 5 | 1.11 | |||
| Am10842 | Acetyl-synthetase | ns | 6 | 1.17 | |||
| Am20064 | T-complex 1 | ns | 9 | 1.19 | |||
| Am20065 | N-myristoyl transferase | ns | 8 | 1.19 | |||
| Am14845 | NAD P-binding | ns | 17 | 1.28 | Orsellinic acid/melleolides [29,30] | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins, C.; Hurley, R.; Almutlaqah, N.; O’Keeffe, G.; Keane, T.M.; Fitzpatrick, D.A.; Owens, R.A. Proteomic Characterization of Armillaria mellea Reveals Oxidative Stress Response Mechanisms and Altered Secondary Metabolism Profiles. Microorganisms 2017, 5, 60. https://doi.org/10.3390/microorganisms5030060
Collins C, Hurley R, Almutlaqah N, O’Keeffe G, Keane TM, Fitzpatrick DA, Owens RA. Proteomic Characterization of Armillaria mellea Reveals Oxidative Stress Response Mechanisms and Altered Secondary Metabolism Profiles. Microorganisms. 2017; 5(3):60. https://doi.org/10.3390/microorganisms5030060
Chicago/Turabian StyleCollins, Cassandra, Rachel Hurley, Nada Almutlaqah, Grainne O’Keeffe, Thomas M. Keane, David A. Fitzpatrick, and Rebecca A. Owens. 2017. "Proteomic Characterization of Armillaria mellea Reveals Oxidative Stress Response Mechanisms and Altered Secondary Metabolism Profiles" Microorganisms 5, no. 3: 60. https://doi.org/10.3390/microorganisms5030060