Prevalence, Distribution, and Phylogeny of Type Two Toxin-Antitoxin Genes Possessed by Cronobacter Species where C. sakazakii Homologs Follow Sequence Type Lineages

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. DNA Extraction for PCR Assay, Microarray, and Whole Genome Sequencing (WGS)
2.3. Microarray Analysis (MA)
2.4. Whole Genome Sequencing (WGS), Assemblies, and Annotation for Comparative Genomic Analysis
2.5. PCR
2.6. Bioinformatics Analysis: A Database and Local BLAST Analysis
3. Results and Discussion
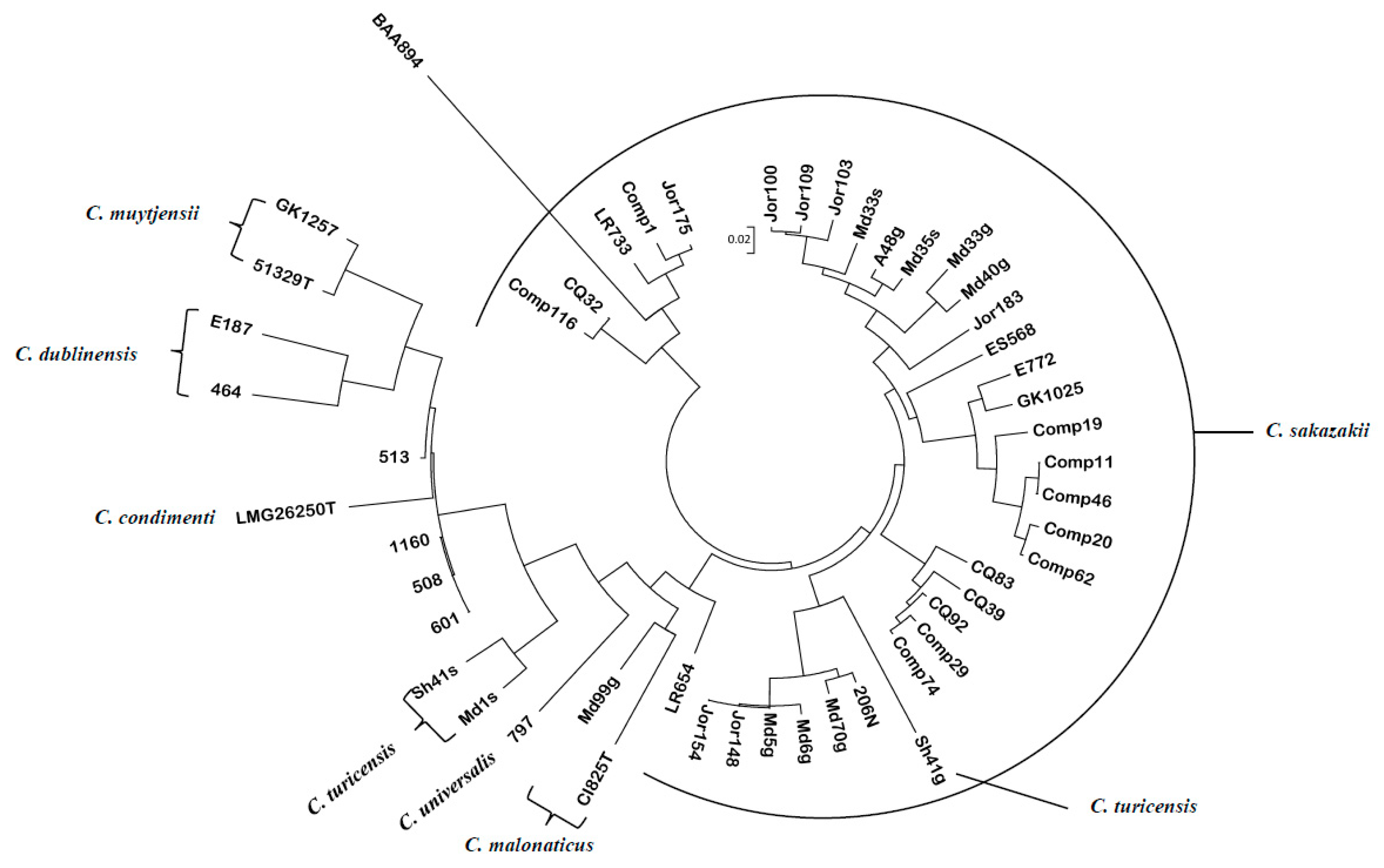
3.1. Pan-Genomic Microarray Analysis Demonstrates That Cronobacter TA Allelic Sequence Divergence Aligned along Species Taxa Lines and Within C. sakazakii Aligned with ST Lineages
3.2. PCR and BLAST Analyses Showed That Not Every C. sakazakii Strain Possessed Similar Numbers or Types of TAs
3.3. Acquisition of TAs Follow Different Evolutionary Incidences Which Draw a Parallel with Occurrence of ST Lineages
3.4. TA Association with Cronobacter Plasmids
3.5. TA Association with Cronobacter Phage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Drèze, P.; Van Melderen, L. Diversity of bacterial type II toxin-antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, Y. Prokaryotic toxin-antitoxin systems: Novel regulations of the toxins. Curr. Genet. 2016, 62, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Rocker, A.; Meinhart, A. Type II toxin: Antitoxin systems. More than small selfish entities? Curr. Genet. 2016, 62, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Hiraga, S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. USA 1983, 80, 4784–4788. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Fernández-García, L.; Blasco, L.; Lopez, M.; Bou, G.; García-Contreras, R.; Wood, T.; Tomas, M. Toxin-antitoxin systems in clinical pathogens. Toxins 2016, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Lehnherr, H.; Maguin, E.; Jafri, S.; Yarmolinsky, M.B. Plasmid addiction genes of bacteriophage P1: Doc, which causes cell death on curing of prophage, and phd, which prevents host death when prophage is retained. J. Mol. Biol. 1993, 233, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, F.; Fraikin, N.; Putrinš, M.; Hallaert, T.; Hauryliuk, V.; Garcia-Pino, A.; Sjödin, A.; Kasvandik, S.; Udekwu, K.; Tenson, T.; et al. Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli type II persister cells. mBio 2018, 9, e00640-18. [Google Scholar] [CrossRef] [PubMed]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the β sliding clamp. Mol. Cell. 2013, 52, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Grim, C.J.; Kotewicz, M.L.; Power, K.A.; Gopinath, G.; Franco, A.A.; Jarvis, K.G.; Yan, Q.Q.; Jackson, S.A.; Sathyamoorthy, V.; Hu, L.; et al. Genomic analysis of the pan genome of the emerging bacterial foodborne pathogen Cronobacter spp. suggests a species-level bidirectional divergence from a conserved genomic core. BMC Genom. 2013, 14, 366. [Google Scholar] [CrossRef] [PubMed]
- Power, K.A.; Yan, Q.; Fox, E.M.; Cooney, S.; Fanning, S. Genome sequence of Cronobacter sakazakii SP291, a persistent thermotolerant isolate derived from a factory producing powdered infant formula. Genome Announc. 2013, 1, e00082-13. [Google Scholar] [CrossRef] [PubMed]
- Iversen, C.; Mullane, N.; McCardell, B.; Tall, B.D.; Lehner, A.; Fanning, S.; Stephan, R.; Joosten, H. Cronobacter gen. nov., a new genus to accommodate the biogroups of Enterobacter sakazakii, and proposal of Cronobacter sakazakii gen. nov., comb. nov., Cronobacter malonaticus sp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp. nov., Cronobacter dublinensis sp. nov., Cronobacter genomospecies 1, and of three subspecies, Cronobacter dublinensis subsp. dublinensis subsp. nov., Cronobacter dublinensis subsp. lausannensis subsp. nov. and Cronobacter dublinensis subsp. lactaridi subsp. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 1442–1447. [Google Scholar] [PubMed]
- Joseph, S.; Cetinkaya, E.; Drahovska, H.; Levican, A.; Figueras, M.J.; Forsythe, S.J. Cronobacter condimenti sp. nov., isolated from spiced meat, and Cronobacter universalis sp. nov., a species designation for Cronobacter sp. genomospecies 1, recovered from a leg infection, water and food ingredients. Int. J. Syst. Evol. Microbiol. 2011, 62, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Stephan, R.; Lehner, A.; Tischler, P.; Rattei, T. Complete genome sequence of Cronobacter turicensis LMG 23827, a food-borne pathogen causing deaths in neonates. J. Bacteriol. 2011, 193, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.B.; Braden, C.R. Invasive Enterobacter sakazakii disease in infants. Emerg. Infect. Dis. 2006, 12, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.K. Enterobacter sakazakii infections among neonates, infants, children, and adults. Case reports and a review of the literature. Medicine 2001, 80, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Jason, J. Prevention of invasive Cronobacter infections in young infants fed powdered infant formulas. Pediatrics 2012, 130, e1076–e1084. [Google Scholar] [CrossRef] [PubMed]
- Patrick, M.E.; Mahon, B.E.; Greene, S.A.; Rounds, J.; Cronquist, A.; Wymore, K.; Boothe, E.; Lathrop, S.; Palmer, A.; Bowen, A. Incidence of Cronobacter spp. infections, United States, 2003–2009. Emerg. Infect. Dis. 2014, 20, 1520–1523. [Google Scholar] [CrossRef] [PubMed]
- Holý, O.; Petrželová, J.; Hanulík, V.; Chromá, M.; Matoušková, I.; Forsythe, S.J. Epidemiology of Cronobacter spp. isolates from patients admitted to the Olomouc University Hospital (Czech Republic). Epidemiol. Mikrobiol. Imunol. 2014, 63, 69–72. [Google Scholar] [PubMed]
- Gosney, M.A.; Martin, M.V.; Wright, A.E.; Gallagher, M. Enterobacter sakazakii in the mouths of stroke patients and its association with aspiration pneumonia. Eur. J. Intern. Med. 2006, 17, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Alsonosi, A.; Hariri, S.; Kajsík, M.; Oriešková, M.; Hanulík, V.; Röderová, M.; Petrželová, J.; Kollárová, H.; Drahovská, H.; Forsythe, S. The speciation and genotyping of Cronobacter isolates from hospitalized patients. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1979–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.Q.; Condell, O.; Power, K.; Butler, F.; Tall, B.D.; Fanning, S. Cronobacter species (formerly known as Enterobacter sakazakii) in powdered infant formula: A review of our current understanding of the biology of this bacterium. J. Appl. Microbiol. 2012, 113, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Himelright, I.; Harris, E.; Lorch, V.; Anderson, M.; Jones, T.; Craig, A.; Kuehnert, M.; Forster, T.; Arduino, M.; Jensen, B.; et al. Enterobacter sakazakii infections associated with the use of powdered infant formula—Tennessee, 2001. Morb. Mortal. Wkly. Rep. 2002, 51, 297–300. [Google Scholar]
- Tall, B.D.; Chen, Y.; Yan, Q.Q.; Gopinath, G.R.; Grim, C.J.; Jarvis, K.G.; Fanning, S.; Lampel, K.A. Cronobacter: An emergent pathogen using meningitis to neonates through their feeds. Sci. Prog. 2014, 97, 154–172. [Google Scholar] [PubMed]
- Noriega, F.R.; Kotloff, K.L.; Martin, M.A.; Schwalbe, R.S. Nosocomial bacteremia caused by Enterobacter sakazakii and Leuconostoc mesenteroides resulting from extrinsic contamination of infant formula. Pediatr. Infect. Dis. J. 1990, 9, 447–449. [Google Scholar] [PubMed]
- Friedemann, M. Enterobacter sakazakii in food and beverages (other than infant formula and milk powder). Int. J. Food Microbiol. 2007, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.; Wiesenfeld, H.C.; Kloesz, J.L.; Pasculle, A.W.; Nowalk, A.J.; Brink, L.; Elliot, E.; Martin, H.; Tarr, C.L. Notes from the Field: Cronobacter sakazakii infection associated with feeding extrinsically contaminated expressed human milk to a premature infant—Pennsylvania. Morb. Mortal. Wkly. Rep. 2017, 66, 761–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullan, R.; Menon, V.; Beukers, A.G.; Jensen, S.O.; van Hal, S.J.; Davis, R. Cronobacter sakazakii infection from expressed breast milk, Australia. Emerg. Infect. Dis. 2018, 24, 393–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sharoud, W.M.; El-Din, M.Z.; Ziada, D.M.; Ahmed, S.F.; Klena, J.D. Surveillance and genotyping of Enterobacter sakazakii suggest its potential transmission from milk powder into imitation recombined soft cheese. J. Appl. Microbiol. 2008, 105, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Osaili, T.M.; Shaker, R.R.; Al-Haddaq, M.S.; Al-Nabulsi, A.A.; Holley, R.A. Heat resistance of Cronobacter species (Enterobacter sakazakii) in milk and special feeding formula. J. Appl. Microbiol. 2009, 107, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Tall, B.D.; Gopinath, G.R.; Gangiredla, J.; Patel, I.R.; Fanning, S.; Lehner, A. Food Microbiology: Fundamentals and Frontiers, 5th ed.; Doyle, M.P., Diez-Gonzalez, F., Hill, C., Eds.; ASM Press: Washington, DC, USA, 2019; pp. 389–414. [Google Scholar]
- Gopinath, G.R.; Chase, H.R.; Gangiredla, J.; Eshwar, A.; Jang, H.; Patel, I.; Negrete, F.; Finkelstein, S.; Park, E.; Chung, T.; et al. Genomic characterization of malonate positive Cronobacter sakazakii serotype O:2, sequence type 64 strains, isolated from clinical, food, and environment samples. Gut Pathog. 2018, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthold-Pluta, A.; Garbowska, M.; Stefanska, I.; Pluta, A. Microbiological quality of selected ready-to-eat leaf vegetables, sprouts and non-pasteurized fresh fruit-vegetable juices including the presence of Cronobacter spp. Food Microbiol. 2017, 65, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Vasconcellos, L.; Carvalho, C.T.; Tavares, R.O.; de Mello Medeiros, V.; de Oliveira Rosas, C.; Silva, J.N.; Dos Reis Lopes, S.M.; Forsythe, S.J.; Brandão, M.L.L. Isolation, molecular and phenotypic characterization of Cronobacter spp. in ready-to-eat salads and foods from Japanese cuisine commercialized in Brazil. Food Res. Int. 2018, 107, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Pava-Ripoll, M.; Pearson, R.E.; Miller, A.K.; Ziobro, G.C. Prevalence and relative risk of Cronobacter spp., Salmonella spp., and Listeria monocytogenes associated with the body surfaces and guts of individual filth flies. Appl. Environ. Microbiol. 2012, 78, 7891–7902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehner, A.; Tall, B.D.; Fanning, S. Cronobacter spp.-opportunistic foodborne pathogens: An update on evolution, osmotic adaptation and pathophysiology. Curr. Clin. Microbiol. Rep. 2017, 5, 97–105. [Google Scholar] [CrossRef]
- Cruz, A.; Xicohtencatl-Cortes, J.; Gonzalez-Pedrajo, B.; Bobadilla, M.; Eslava, C.; Rosas, I. Virulence traits in Cronobacter species isolated from different sources. Can. J. Microbiol. 2011, 57, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Chase, H.R.; Gangiredla, J.; Gopinath, G.R.; Grim, C.J.; Patel, I.R.; Kothary, M.H.; Jackson, S.A.; Mammel, M.K.; Carter, L.; et al. Analysis of the molecular diversity among Cronobacter species isolated from filth flies using a pan genomic DNA microarray and whole genome sequencing. Front. Microbiol. 2019. submitted. [Google Scholar]
- Umeda, N.S.; De Filippis, I.; Forsythe, S.J.; Brandão, M.L.L. Phenotypic characterization of Cronobacter spp. strains isolated from foods and clinical specimens in Brazil. Food Res. Int. 2017, 102, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Addy, N.; Ewing, L.; Beaubrun, J.J.G.; Lee, Y.; Woo, J.; Negrete, F.; Finkelstein, S.; Tall, B.D.; Lehner, A.; et al. Whole Genome Sequences of Cronobacter sakazakii isolates obtained from plant-origin foods and dried food manufacturing environments. Genome Announc. 2018, 6, e00223-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.; Gopinath, G.R.; Chase, H.R.; Gangiredla, J.; Eshwar, A.; Patel, I.; Addy, N.; Ewing, L.; Beaubrunm, J.J.G.; Negrete, F.; et al. Draft genomes of Cronobacter sakazakii strains isolated from dried spices bring unique insights into the epidemiology of plant-associated strains. Stand. Genom. Sci. 2018, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Sani, N.A.; Odeyemi, O.A. Occurrence and prevalence of Cronobacter spp. in plant and animal derived food sources: A systematic review and meta-analysis. SpringerPlus 2015, 4, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, W.; Guo, B.; Shi, X.; Cheng, T.; Chen, M.; Jiang, X.; Ye, Y.; Wang, J.; Xie, G.; Ding, J. An investigation of an acute gastroenteritis outbreak: Cronobacter sakazakii, a potential cause of food-borne illness. Front. Microbiol. 2018, 9, 2549. [Google Scholar] [CrossRef] [PubMed]
- Restaino, L.; Frampton, E.W.; Lionberg, W.C.; Becker, R.J. A chromogenic plating medium for the isolation and identification of Enterobacter sakazakii from foods, food ingredients, and environmental sources. J. Food Prot. 2006, 69, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Jaradat, Z.W.; Ababneh, Q.O.; Saadoun, I.M.; Samara, N.A.; Rashdan, A.M. Isolation of Cronobacter spp. (formerly Enterobacter sakazakii) from infant food, herbs and environmental samples and the subsequent identification and confirmation of the isolates using biochemical, chromogenic assays, PCR and 16S rRNA sequencing. BMC Microbiol. 2009, 9, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chon, J.W.; Song, K.Y.; Kim, S.Y.; Hyeon, J.Y.; Seo, K.H. Isolation and characterization of Cronobacter from desiccated foods in Korea. J. Food Sci. 2012, 77, M354–M358. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wang, J.; Gangiredla, J.; Cao, Y.; Martins, M.; Gopinath, G.R.; Stephan, R.; Lampel, K.; Tall, B.D.; Fanning, S. Comparative genotypic and phenotypic analysis of Cronobacter Species cultured from four powdered infant formula production facilities: Indication of pathoadaptation along the food chain. Appl. Environ. Microbiol. 2015, 81, 4388–4402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, H.R.; Gopinath, G.R.; Eshwar, A.K.; Stoller, A.; Fricker-Feer, C.; Gangiredla, J.; Patel, I.R.; Cinar, H.N.; Jeong, H.; Lee, C.; et al. Comparative genomic characterization of the highly persistent and potentially virulent Cronobacter sakazakii ST83, CC65 strain H322 and other ST83 strains. Front. Microbiol. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Stoop, B.; Lehner, A.; Iversen, C.; Fanning, S.; Stephan, R. Development and evaluation of rpoB based PCR systems to differentiate the six proposed species within the genus Cronobacter. Int. J. Food Microbiol. 2009, 136, 165–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehner, A.; Fricker-Feer, C.; Stephan, R. Identification of the recently described Cronobacter condimenti by a rpoB based PCR system. J. Med. Microbiol. 2012, 61, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.; Lindsey, L.A.; Grim, C.J.; Sathyamoorthy, V.; Jarvis, K.G.; Gopinath, G.; Lee, C.; Sadowski, J.A.; Trach, L.; Pava-Ripoll, M.; et al. Multiplex PCR assay targeting a diguanylate cyclase-encoding gene, cgcA, to differentiate species within the genus Cronobacter. Appl. Environ. Microbiol. 2013, 79, 734–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, S.; Sonbol, H.; Hariri, S.; Desai, P.; McClelland, M.; Forsythe, S.J. Diversity of the Cronobacter genus as revealed by multilocus sequence typing. J. Clin. Microbiol. 2012, 50, 3031–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.; Loughlin, M.; Caubilla-Barron, J.; Kucerova, E.; Manning, G.; Dowson, C.; Forsythe, S. Multilocus sequence typing of Cronobacter sakazakii and Cronobacter malonaticus reveals stable clonal structures with clinical significance which do not correlate with biotypes. BMC Microbiol. 2009, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tall, B.D.; Gangiredla, J.; Gopinath, G.R.; Yan, Q.Q.; Chase, H.R.; Lee, B.; Hwang, S.; Trach, L.; Park, E.; Yoo, Y.J.; et al. Development of a custom-designed, pan genomic DNA microarray to characterize strain-level diversity among Cronobacter spp. Front. Pediatr. 2015, 3, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamruzzaman, M.; Iredell, J. A ParDE-family toxin antitoxin system in major resistance plasmids of Enterobacteriaceae confers antibiotic and heat tolerance. Sci. Rep. 2019, 9, 9872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Jolley, K.A.; Maiden, M.C.J. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrete, F.; Gangiredla, J.; Jang, H.; Chase, H.R.; Woo, J.; Lee, Y.; Patel, I.; Finkelstein, S.B.; Tall, B.D.; Gopinath, G.R. Prevalence and distribution of efflux pump complexes genes in Cronobacter sakazakii using whole genome and pan-genomic datasets. Curr. Opin. Food Sci. 2019, 30, 32–42. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Thomas, R.H., Ed.; Oxford University Press: Oxford, UK, 2000; p. 333. [Google Scholar]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.A.; Hu, L.; Grim, C.J.; Gopinath, G.; Sathyamoorthy, V.; Jarvis, K.G.; Lee, C.; Sadowski, J.; Kim, J.; Kothary, M.H.; et al. Characterization of putative virulence genes on the related RepFIB plasmids harbored by Cronobacter spp. Appl. Environ. Microbiol. 2011, 77, 3255–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucerova, E.; Clifton, S.W.; Xia, X.Q.; Long, F.; Porwollik, S.; Fulton, L.; Fronick, C.; Minx, P.; Kyung, K.; Warren, W.; et al. Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS ONE 2010, 5, e9556. [Google Scholar] [CrossRef] [PubMed]
- Moine, D.; Kassam, M.; Baert, L.; Tang, Y.; Barretto, C.; Ngom Bru, C.; Klijn, A.; Descombes, P. Fully closed genome sequences of five type strains of the genus Cronobacter and one Cronobacter sakazakii Strain. Genome Announc. 2016, 4, e00142-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Species ID | ST a, CC b | Source | Country | NCBI Accession No. |
|---|---|---|---|---|---|
| Comp1 | C. sakazakii | ST1, CC1 | Environment, dairy powder manufacturing facility | USA | WAGE00000000 |
| Comp11 | C. sakazakii | ST64, CC64 | Environment, dairy powder manufacturing facility | USA | NHQL01000000 |
| Comp19 | C. sakazakii | ST64, CC64 | Environment, dairy powder manufacturing facility | USA | NHQM00000000 |
| Comp20 | C. sakazakii | ST64, CC64 | Environment, dairy powder manufacturing facility | USA | NEXY00000000 |
| Comp29 | C. sakazakii | ST40, CC40 | Environment, dairy powder manufacturing facility | USA | Not available |
| Comp46 | C. sakazakii | ST64, CC64 | Environment, dairy powder manufacturing facility | USA | NEYA00000000 |
| Comp62 | C. sakazakii | ST1, CC1 | Environment, dairy powder manufacturing facility | USA | WAGM00000000 |
| Comp74 | C. sakazakii | ST1, CC1 | Environment, dairy powder manufacturing facility | USA | Not available |
| Comp116 | C. sakazakii | ST1, CC1 | Environment, dairy powder manufacturing facility | USA | WAGR00000000 |
| Jor175 | C. sakazakii | ST1, CC1 | Food, spices | Jordan | NITO00000000 |
| LR733 | C. sakazakii | ST1, CC1 | Food, organic flour | USA | PTOU00000000 |
| CQ32 | C. sakazakii | ST1, CC1 | Environment, powdered infant formula facility | Ireland | WAGV00000000 |
| BAA-894 | C. sakazakii | ST1, CC1 | Food, powdered infant formula | USA | CP000783 |
| Jor100 | C. sakazakii | ST643, CC13 | Food, semolina | Jordan | NITS01000000 |
| Jor103 | C. sakazakii | ST643, CC13 | Food, spices | Jordan | NITR00000000 |
| Jor109 | C. sakazakii | ST643, CC13 | Food, grapes | Jordan | NITQ00000000 |
| Jor148 | C. sakazakii | ST4, CC4 | Food, spices | Jordan | PVCF00000000 |
| Jor154 | C. sakazakii | ST4, CC4 | Food, spices | Jordan | NITP00000000 |
| Jor183 | C. sakazakii | ST21, CC21 | Food, spices | Jordan | NITN00000000 |
| CQ39 | C. sakazakii | ST40, CC40 | Environment, powdered infant formula facility | Ireland | WAGW00000000 |
| CQ83 | C. sakazakii | ST40, CC40 | Environment, powdered infant formula facility | Ireland | Not available |
| CQ92 | C. sakazakii | ST40, CC40 | Environment, powdered infant formula facility | Ireland | Not available |
| GK1025 | C. sakazakii | ST64, CC64 | Environment, powdered infant formula facility | Germany | MCOE00000000 |
| Md33s | C. sakazakii | ST8, CC8 | Fly, Musca domestica, surface | USA | MRXC00000000 |
| Md33g | C. sakazakii | ST8, CC8 | Fly, Musca domestica, gut | USA | MSAI00000000 |
| Md35s | C. sakazakii | ST8, CC8 | Fly, Musca domestica, surface | USA | MRXD00000000 |
| Md40g | C. sakazakii | ST8, CC8 | Fly, Musca domestica, gut | USA | MRXE00000000 |
| Anth48g | C. sakazakii | ST221 | Fly, Anthomyiidae spp., gut | USA | MRXF00000000 |
| Md5s | C. sakazakii | ST4, CC4 | Fly, Musca domestica, surface | USA | MRWZ00000000 |
| Md6g | C. sakazakii | ST4, CC4 | Fly, Musca domestica, gut | USA | MRXB00000000 |
| Md70g | C. sakazakii | ST4, CC4 | Fly, Musca domestica, gut | USA | MRXG00000000 |
| Md1g | C. sakazakii | ST4, CC4 | Fly, Musca domestica, gut | USA | MSAH00000000 |
| 206N | C. sakazakii | ST4, CC4 | Clinical | Ireland | WAEU00000000 |
| ES568 | C. sakazakii | ST3, CC3 | Environment, powdered infant formula facility | Switzerland | Not available |
| E654 | C. sakazakii | ST1, CC1 | Clinical | Ireland | NCWF00000000 |
| E772 | C. sakazakii | ST64, CC64 | Food, milk powder | France | NHQS00000000 |
| GK1257 | C. muytjensii | ST546 | Environment, powdered infant formula facility | Germany | WAGD00000000 |
| Md99g | C. malonaticus | ST60 | Fly, Musca domestica, gut | USA | MSAF00000000 |
| Md1sN | C. turicensis | ST519 | Fly, Musca domestica, gut | USA | VOEL00000000 |
| Sh41s | C. turicensis | ST569 | Fly, Sarcophaga haemorrhoidalis, surface | USA | MSAG01000000 |
| Sh41g | C. turicensis | ST569 | Fly, Sarcophaga haemorrhoidalis, gut | USA | MRZS00000000 |
| NCTC 9529T (797) | C. universalis | ST54 | Environment, water | UK | NZ_CP012257 |
| 51329T | C. muytjensii | ST81 | Unknown | USA | NZ_CP012268 |
| 187 (LMG23823T) | C. dublinensis | ST106 | Environment, powdered infant formula facility | Ireland | NZ_CP012266 |
| 1330 (LMG 26250T) | C. condimenti | ST98 | Food, spiced sausage | Slovakia | NZ_CP012264 |
| 464 (LMG23825T) | C. dublinensis | ST79 | Environment, milk powder production facility | Zimbabwe | AJKX00000000 |
| CI825 (LMG23826T) | C. malonaticus | ST7 | Clinical, breast abscess | USA | NZ_CP013940 |
| 508 (LMG 23730T) | Siccibacter turicensis | N/A c | Food, fruit powder | Switzerland | AWFZ01000000 |
| 601 (LMG 24057T) | Franconibacter pulveris | N/A | Food, fruit powder | Switzerland | AXSY00000000 |
| 1160 (LMG 24058T) | Franconibacter pulveris | N/A | Food, fruit powder | Switzerland | AXSZ00000000 |
| 513 (LMG 23732t) | Franconibacter helveticus | N/A | Food, fruit powder | Switzerland | AXDK00000000 |
| Primer Target a | Forward and Reverse Primer | Sequence (5′-3′) | Amplicon Size |
|---|---|---|---|
| ESA00258 | 00258F5 | CGA GAC CGT TAA AGC GCA AT | 211 bp |
| 00258R3 | CCC CTG GTA TAC GGT CAG GT | ||
| ESA00804 | 00804F10 | TGG AGA TCA GAT GGA CGA AGC | 251 bp |
| 00804R9 | TGT GGT TGT CGT TCT GCG TT | ||
| ESA01887 | 01887F2 | TCA GGC ATA AAG GCC TGC AA | 239 bp |
| 01887R7 | AAA GAC ATC GCC ATC CCG AA | ||
| ESA03838 | 03838F3 | AAT TTT TCA TCC GGT CGC GG | 301 bp |
| 03838R4 | ATG GCT GAG CTC CTC CAA TC | ||
| ESA04372 | 04372F7 | GCG CGA CCC TTA TTT CTG GT | 538 bp |
| 04372R1 | TTT TCT CAA GCG GTG CCA GA |
| Strain | Toxin PCR Patterns Observed among 22 C. sakazakii Strains | ||||
|---|---|---|---|---|---|
| ESA_00258 | ESA_00804 | ESA_01887 | ESA_03838 | ESA_04372 | |
| Comp 11 | + | + | + | − | + |
| Comp 13 | + | + | + | − | + |
| Comp 14 | + | + | + | − | + |
| Comp 15 | + | + | + | − | + |
| Comp 18 | + | + | + | − | + |
| Comp 19 | + | + | + | − | + |
| Comp 20 | + | + | + | − | + |
| Comp 26 | + | − | + | + | + |
| Comp 28 | − | + | − | − | − |
| Comp 42 | + | + | + | − | + |
| Comp 45 | + | + | + | − | + |
| Comp 46 | + | + | + | − | + |
| Comp 48 | + | + | + | − | + |
| Comp 49 | − | + | + | − | + |
| Comp 52 | + | + | + | − | + |
| Comp 53 | + | + | + | − | + |
| Comp 54 | + | + | + | − | + |
| Comp 55 | − | + | − | − | + |
| Comp 57 | + | + | + | − | + |
| Comp 58 | + | + | + | − | + |
| Comp 59 | + | + | + | − | + |
| Comp 60 | + | + | + | − | + |
| Toxin (T) or Antitoxin (A) | Toxin Gene | NCBI Annotations | Presence in C. saks (%) |
|---|---|---|---|
| A | ESA_00257 | RelB protein (antitoxin to RelE) | 83 |
| T | ESA_00258 | RelE antibacterial toxin protein | 88 |
| A | ESA_00803 | transcriptional regulator2C XRE family | 100 |
| T | ESA_00804 | hypothetical protein | 100 |
| T | ESA_00912 | FIG00554131: hypothetical protein | 13 |
| A | ESA_00913 | FIG00553297: hypothetical protein | 38 |
| T | ESA_01146 | hypothetical protein | 18 |
| A | ESA_01147 | Putative transcriptional regulator | 94 |
| A | ESA_01886 | HigA protein (antitoxin to HigB) | 37 |
| T | ESA_01887 | HigB toxin protein | 100 |
| A | ESA_02142 | HigA protein (antitoxin to HigB) | 75 |
| T | ESA_02143 | HigB toxin protein | 21 |
| A | ESA_03837 | FIG00554128: hypothetical protein | 76 |
| T | ESA_03838 | FIG00554128: hypothetical protein | 77 |
| A | ESA_03866 | Putative merR family bacterial regulatory protein | 29 |
| T | ESA_03867 | FIG00642734: hypothetical protein | 13 |
| A | ESA_04288 | Prevent host death protein2C Phd antitoxin | 14 |
| T | ESA_04289 | Death on curing protein2C Doc toxin | 13 |
| A | ESA_04371 | FIG00553654: hypothetical protein | 58 |
| T | ESA_04372 | Cell filamentation protein fic | 91 |
| T | ESA3p05543 | hypothetical_protein | 14 |
| A | ESA3p05544 | hypothetical_protein | 23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finkelstein, S.; Negrete, F.; Jang, H.; Gangiredla, J.; Mammel, M.; Patel, I.R.; Chase, H.R.; Woo, J.; Lee, Y.; Wang, C.Z.; et al. Prevalence, Distribution, and Phylogeny of Type Two Toxin-Antitoxin Genes Possessed by Cronobacter Species where C. sakazakii Homologs Follow Sequence Type Lineages. Microorganisms 2019, 7, 554. https://doi.org/10.3390/microorganisms7110554
Finkelstein S, Negrete F, Jang H, Gangiredla J, Mammel M, Patel IR, Chase HR, Woo J, Lee Y, Wang CZ, et al. Prevalence, Distribution, and Phylogeny of Type Two Toxin-Antitoxin Genes Possessed by Cronobacter Species where C. sakazakii Homologs Follow Sequence Type Lineages. Microorganisms. 2019; 7(11):554. https://doi.org/10.3390/microorganisms7110554
Chicago/Turabian StyleFinkelstein, Samantha, Flavia Negrete, Hyein Jang, Jayanthi Gangiredla, Mark Mammel, Isha R. Patel, Hannah R. Chase, JungHa Woo, YouYoung Lee, Caroline Z. Wang, and et al. 2019. "Prevalence, Distribution, and Phylogeny of Type Two Toxin-Antitoxin Genes Possessed by Cronobacter Species where C. sakazakii Homologs Follow Sequence Type Lineages" Microorganisms 7, no. 11: 554. https://doi.org/10.3390/microorganisms7110554