High-Level Production of Bacteriotoxic Phospholipase A1 in Bacterial Host Pseudomonas fluorescens via ABC Transporter-Mediated Secretion and Inducible Expression

 ,
,
Abstract
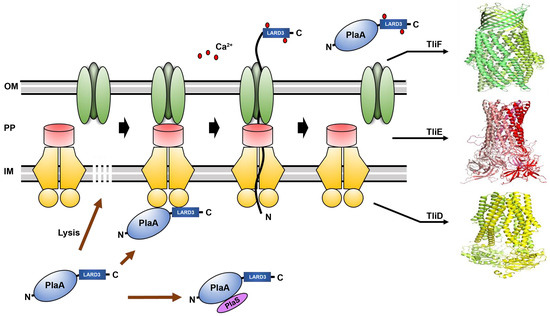
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Growth Conditions
2.2. Construction of Plasmids
2.3. Construction of P. fluorescens ZYAI and P. fluorescens AlgD::LacI
2.4. Analyses of PlaA Expression and Fermentation
2.5. Measurement of Secretory PlaA Activity
2.6. Degumming of Crude Plant Oil and Lecithin Hydrolysis
3. Results
3.1. PlaA Expression in E. coli
3.2. Construction of P. fluorescens ZYAI
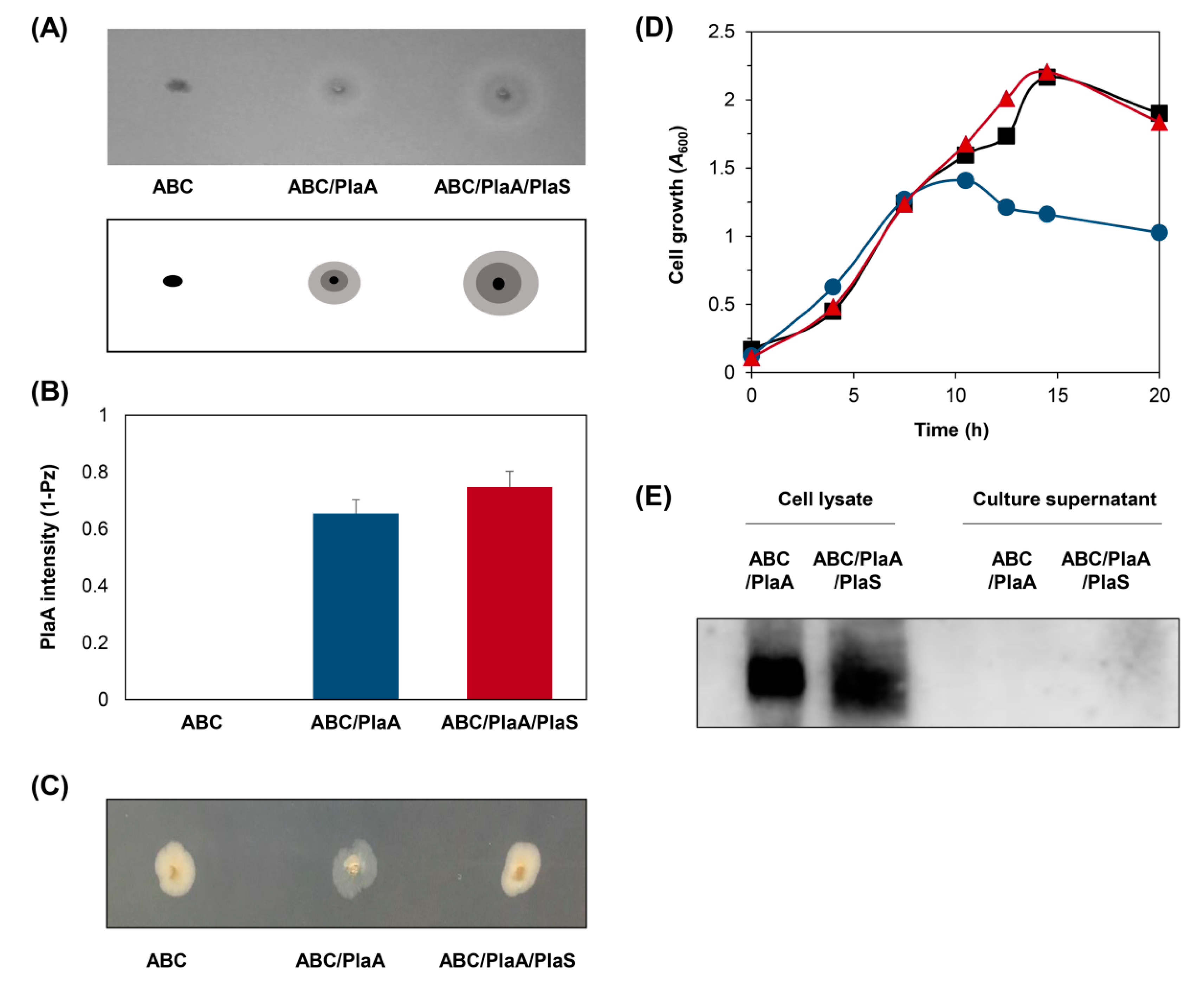
3.3. Secretion of PlaA in P. fluorescens ZYAI
3.4. Production of PlaA in Fermenter
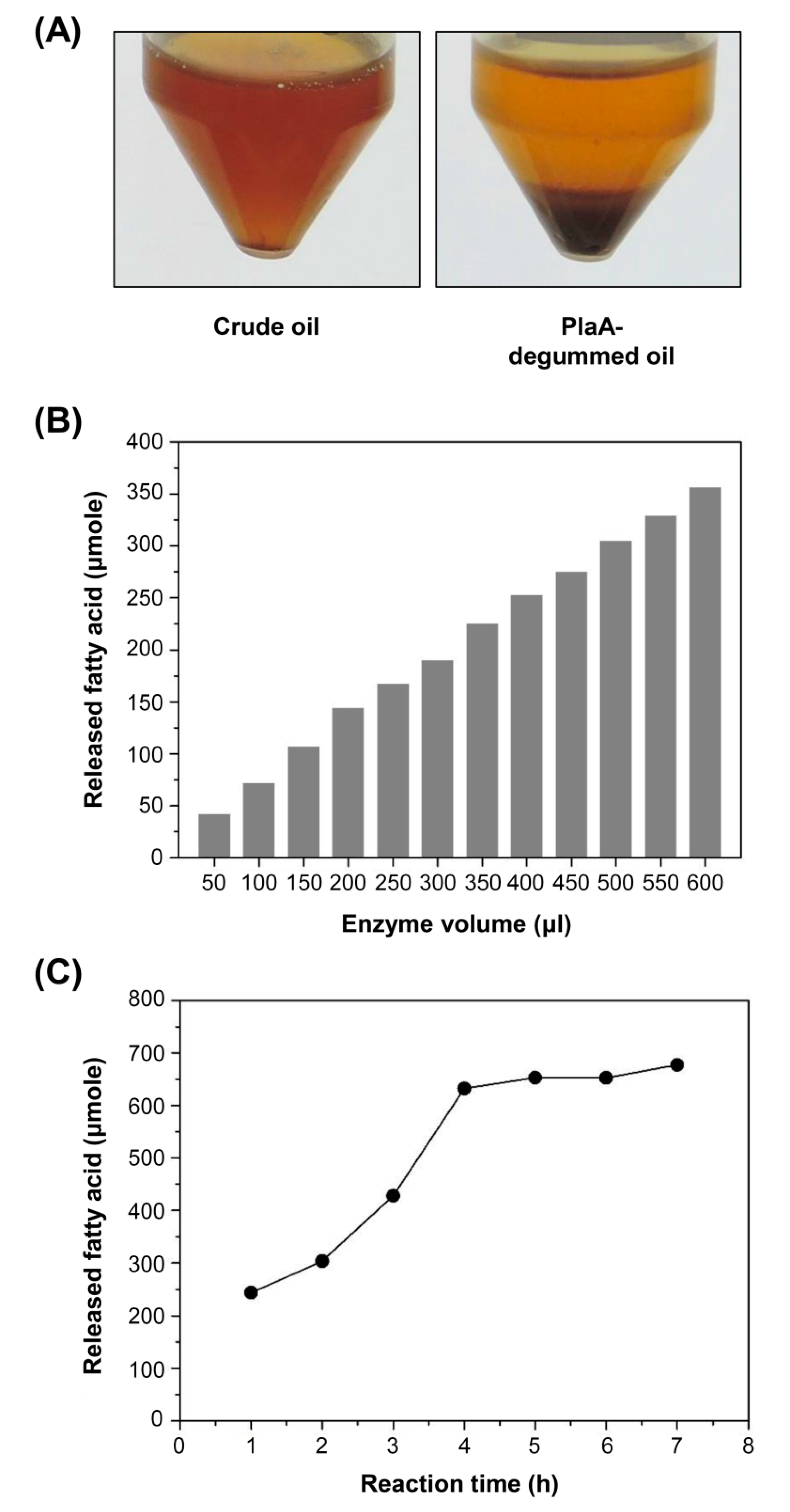
3.5. Use of Secreted PlaA in Degumming Crude Plant Oil and Hydrolysis of Lecithin
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richmond, G.S.; Smith, T.K. Phospholipases A(1). Int. J. Mol. Sci. 2011, 12, 588–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.K.; Kim, M.K.; Rhee, J.S. Cloning and expression of the gene encoding phospholipase A1 from Serratia sp. MK1 in Escherichia coli. J. Biotechnol. 1999, 72, 103–114. [Google Scholar] [CrossRef]
- Shiba, Y.; Ono, C.; Fukui, F.; Watanabe, I.; Serizawa, N.; Gomi, K.; Yoshikawa, H. High-Level Secretory Production of Phospholipase A1 by Saccharomyces cerevisiae and Aspergillus oryzae. Biosci. Biotechnol. Biochem. 2001, 65, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, N.; Tommassen, J.; Lustig, A.; Rosenbusch, J.P.; Verheij, H.M. Dimerization regulates the enzymatic activity of Escherichia coli outer membrane phospholipase A. J. Biol. Chem. 1997, 272, 3179–3184. [Google Scholar] [CrossRef] [Green Version]
- Givskov, M.; Molin, S. Secretion of Serratia liquefaciens phospholipase from Escherichia coli. Mol. Microbiol. 1993, 8, 229–242. [Google Scholar] [CrossRef]
- Yang, B.; Wang, Y.-H.; Yang, J.-G. Optimization of enzymatic degumming process for rapeseed oil. J. Am. Oil Chem. Soc. 2006, 83, 653–658. [Google Scholar] [CrossRef]
- Yang, B.; Zhou, R.; Yang, J.-G.; Wang, Y.-H.; Wang, W.-F. Insight into the enzymatic degumming process of soybean oil. J. Am. Oil Chem. Soc. 2008, 85, 421–425. [Google Scholar] [CrossRef]
- Bang, H.J.; Kim, I.H.; Kim, B.H. Phospholipase A1-catalyzed hydrolysis of soy phosphatidylcholine to prepare l-alpha-glycerylphosphorylcholine in organic-aqueous media. Food Chem. 2016, 190, 201–206. [Google Scholar] [CrossRef]
- Yang, J.-G.; Wang, Y.-H.; Yang, B.; Mainda, G.; Guo, Y. Degumming of vegetable oil by a new microbial lipase. Food Technol. Biotechnol. 2006, 44, 101. [Google Scholar]
- Sheelu, G.; Kavitha, G.; Fadnavis, N.W. Efficient immobilization of lecitase in gelatin hydrogel and degumming of rice bran oil using a spinning basket reactor. J. Am. Oil Chem. Soc. 2008, 85, 739–748. [Google Scholar] [CrossRef]
- Songer, J.G. Bacterial phospholipases and their role in virulence. Trends Microbiol. 1997, 5, 156–161. [Google Scholar] [CrossRef]
- Lim, H.J.; Park, Y.J.; Jang, Y.J.; Choi, J.E.; Oh, J.Y.; Park, J.H.; Song, J.K.; Kim, D.M. Cell-free synthesis of functional phospholipase A1 from Serratia sp. Biotechnol. Biofuels 2016, 9, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, T.D.; Chew, L.; Davis, J.W.; Frawley, N.; Foley, H.H.; Stelman, S.J.; Thomas, J.; Wolt, J.; Hanselman, D.S. Safety evaluation of an alpha-amylase enzyme preparation derived from the archaeal order Thermococcales as expressed in Pseudomonas fluorescens biovar I. Regul. Toxicol. Pharmacol. 2003, 37, 149–168. [Google Scholar] [CrossRef]
- Rainey, P.B.; Bailey, M.J. Physical and genetic map of the Pseudomonas fluorescens SBW25 chromosome. Mol. Microbiol. 1996, 19, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.C.; Jenings, A.F.; Mun, D.M.; McGovern, P.M.; Chew, L.C. Auxotrophic markers pyrF and proC can replace antibiotic markers on protein production plasmids in high-cell-density Pseudomonas fluorescens fermentation. Biotechnol. Prog. 2005, 21, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Squires, C.H.; Retallack, D.M.; Chew, L.C.; Ramseier, T.M.; Schneider, J.C.; Talbot, H.W. Heterologous protein production in P. fluorescens. BioProcesss Int. 2004, 2, e58. [Google Scholar]
- Ryu, J.; Lee, U.; Park, J.; Yoo, D.H.; Ahn, J.H. A vector system for ABC transporter-mediated secretion and purification of recombinant proteins in Pseudomonas species. Appl. Environ. Microbiol. 2015, 81, 1744–1753. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Pan, J.G.; Rhee, J.S. Identification of the tliDEF ABC transporter specific for lipase in Pseudomonas fluorescens SIK W1. J. Bacteriol. 1999, 181, 1847–1852. [Google Scholar] [CrossRef] [Green Version]
- Son, M.; Moon, Y.; Oh, M.J.; Han, S.B.; Park, K.H.; Kim, J.G.; Ahn, J.H. Lipase and protease double-deletion mutant of Pseudomonas fluorescens suitable for extracellular protein production. Appl. Environ. Microbiol. 2012, 78, 8454–8462. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.K.; Kim, J.K.; Rhee, J.S. Isolation of a phospholipase A1-producing microorganism. J. Ind. Microbiol. 1996, 16, 171–174. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Ahn, J.H.; Pan, J.G.; Rhee, J.S. Homologous expression of the lipase and ABC transporter gene cluster, tliDEFA, enhances lipase secretion in Pseudomonas spp. Appl. Environ. Microbiol. 2001, 67, 5506–5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Marco, A.; Deuerling, E.; Mogk, A.; Tomoyasu, T.; Bukau, B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 2007, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, A.; Tauch, A.; Jager, W.; Kalinowski, J.; Thierbach, G.; Puhler, A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 1994, 145, 69–73. [Google Scholar] [CrossRef]
- Chung, C.W.; You, J.; Kim, K.; Moon, Y.; Kim, H.; Ahn, J.H. Export of recombinant proteins in Escherichia coli using ABC transporter with an attached lipase ABC transporter recognition domain (LARD). Microb. Cell Fact. 2009, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothavade, R.J.; Panthaki, M.H. Evaluation of phospholipase activity of Candida albicans and its correlation with pathogenicity in mice. J. Med. Microbiol. 1998, 47, 99–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.F.; Wilkinson, I.D.; Gentry, L.O. Plate method for detection of phospholipase activity in Candida albicans. Sabouraudia J. Med. Vet. Mycol. 1982, 20, 7–14. [Google Scholar] [CrossRef]
- Vassar, R.J.; Potel, M.J.; Josephs, R. Studies of the fiber to crystal transition of sickle cell hemoglobin in acidic polyethylene glycol. J. Mol. Biol. 1982, 157, 395–412. [Google Scholar] [CrossRef]
- Reynolds, L.J.; Washburn, W.N.; Deems, R.A.; Dennis, E.A. Assay strategies and methods for phospholipases. Methods Enzymol. 1991, 197, 3–23. [Google Scholar]
- Keen, N.T.; Tamaki, S.; Kobayashi, D.; Trollinger, D. Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 1988, 70, 191–197. [Google Scholar] [CrossRef]
- Chrisope, G.L.; Fox, C.W.; Marshall, R.T. Lecithin agar for detection of microbial phospholipases. Appl. Environ. Microbiol. 1976, 31, 784–786. [Google Scholar] [CrossRef] [Green Version]
- Gimmestad, M.; Sletta, H.; Ertesvag, H.; Bakkevig, K.; Jain, S.; Suh, S.J.; Skjak-Braek, G.; Ellingsen, T.E.; Ohman, D.E.; Valla, S. The Pseudomonas fluorescens AlgG protein, but not its mannuronan C-5-epimerase activity, is needed for alginate polymer formation. J. Bacteriol. 2003, 185, 3515–3523. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, D.J.; Wyckoff, T.J.; Starkey, M.; Keyser, R.; Azadi, P.; O’Toole, G.A.; Parsek, M.R. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. USA 2003, 100, 7907–7912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehm, B.H. Bacterial polymers: biosynthesis, modifications and applications. Nat. Rev. Microbiol. 2010, 8, 578–592. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Song, J.K.; Rhee, J.S. Simultaneous enhancement of thermostability and catalytic activity of phospholipase A(1) by evolutionary molecular engineering. Appl. Environ. Microbiol. 2000, 66, 890–894. [Google Scholar] [CrossRef] [Green Version]
- Song, J.K.; Rhee, J.S. Enhancement of stability and activity of phospholipase A(1) in organic solvents by directed evolution. Biochim. Biophys. Acta 2001, 1547, 370–378. [Google Scholar] [CrossRef]
- Costa, T.R.; Felisberto-Rodrigues, C.; Meir, A.; Prevost, M.S.; Redzej, A.; Trokter, M.; Waksman, G. Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 2015, 13, 343–359. [Google Scholar] [CrossRef]
- Castelli, M.E.; Véscovi, E.G. The Rcs signal transduction pathway is triggered by enterobacterial common antigen structure alterations in Serratia marcescens. J. Bacteriol. 2011, 193, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Nydam, S.D.; Christensen, J.E.; Konkel, M.E.; Orfe, L.; Friel, P.; Call, D.R. Identification of potential type III secretion proteins via heterologous expression of Vibrio parahaemolyticus DNA. Appl. Environ. Microbiol. 2012, 78, 3492–3494. [Google Scholar] [CrossRef] [Green Version]
- Young, B.M.; Young, G.M. YplA is exported by the Ysc, Ysa, and flagellar type III secretion systems of Yersinia enterocolitica. J. Bacteriol. 2002, 184, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Givskov, M.; Eberl, L.; Christiansen, G.; Benedik, M.J.; Molin, S. Induction of phospholipase- and flagellar synthesis in Serratia liquefaciens is controlled by expression of the flagellar master operon flhD. Mol. Microbiol. 1995, 15, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Meysick, K.C.; Seidman, J.; Falconio, J.R. The Yersinia pseudotuberculosis YplA phospholipase differs in its activity, regulation and secretion from that of the Yersinia enterocolitica YplA. Microb. Pathog. 2009, 47, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Schmiel, D.H.; Young, G.M.; Miller, V.L. The Yersinia enterocolitica Phospholipase Gene yplA Is Part of the Flagellar Regulon. J. Bacteriol. 2000, 182, 2314–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, M.E.; Fedrigo, G.V.; Clementín, A.L.; Ielmini, M.V.; Feldman, M.F.; Véscovi, E.G. Enterobacterial common antigen integrity is a checkpoint for flagellar biogenesis in Serratia marcescens. J. Bacteriol. 2008, 190, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Huang, H.; Meng, K.; Yuan, T.; Yao, B.; Shi, Y.; Ouyang, P. A novel cold-adapted phospholipase A 1 from Serratia sp. xjF1: Gene cloning, expression and characterization. Enzym. Microb. Technol. 2008, 42, 187–194. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, Y.J.; Zhang, Z. Using weakly conserved motifs hidden in secretion signals to identify type-III effectors from bacterial pathogen genomes. PLoS ONE 2013, 8, e56632. [Google Scholar] [CrossRef] [Green Version]
- Givskov, M.; Molin, S. Expression of extracellular phospholipase from Serratia liquefaciens is growth-phase-dependent, catabolite-repressed and regulated by anaerobiosis. Mol. Microbiol. 1992, 6, 1363–1374. [Google Scholar] [CrossRef]
- Anderson, M.T.; Mitchell, L.A.; Mobley, H.L. Cysteine biosynthesis controls Serratia marcescens phospholipase activity. J. Bacteriol. 2017, 199, e00159-17. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.X.; Moore, R.K.; Otsuka, F.; Shimasaki, S. Effect of intracellular interactions on the processing and secretion of bone morphogenetic protein-15 (BMP-15) and growth and differentiation factor-9. Implication of the aberrant ovarian phenotype of BMP-15 mutant sheep. J. Biol. Chem. 2003, 278, 3713–3719. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhuang, F.F.; Mullersman, J.E.; Chen, H.; Robertson, E.J.; Warburton, D.; Liu, Y.H.; Shi, W. BMP4 activation and secretion are negatively regulated by an intracellular gremlin-BMP4 interaction. J. Biol. Chem. 2006, 281, 29349–29356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
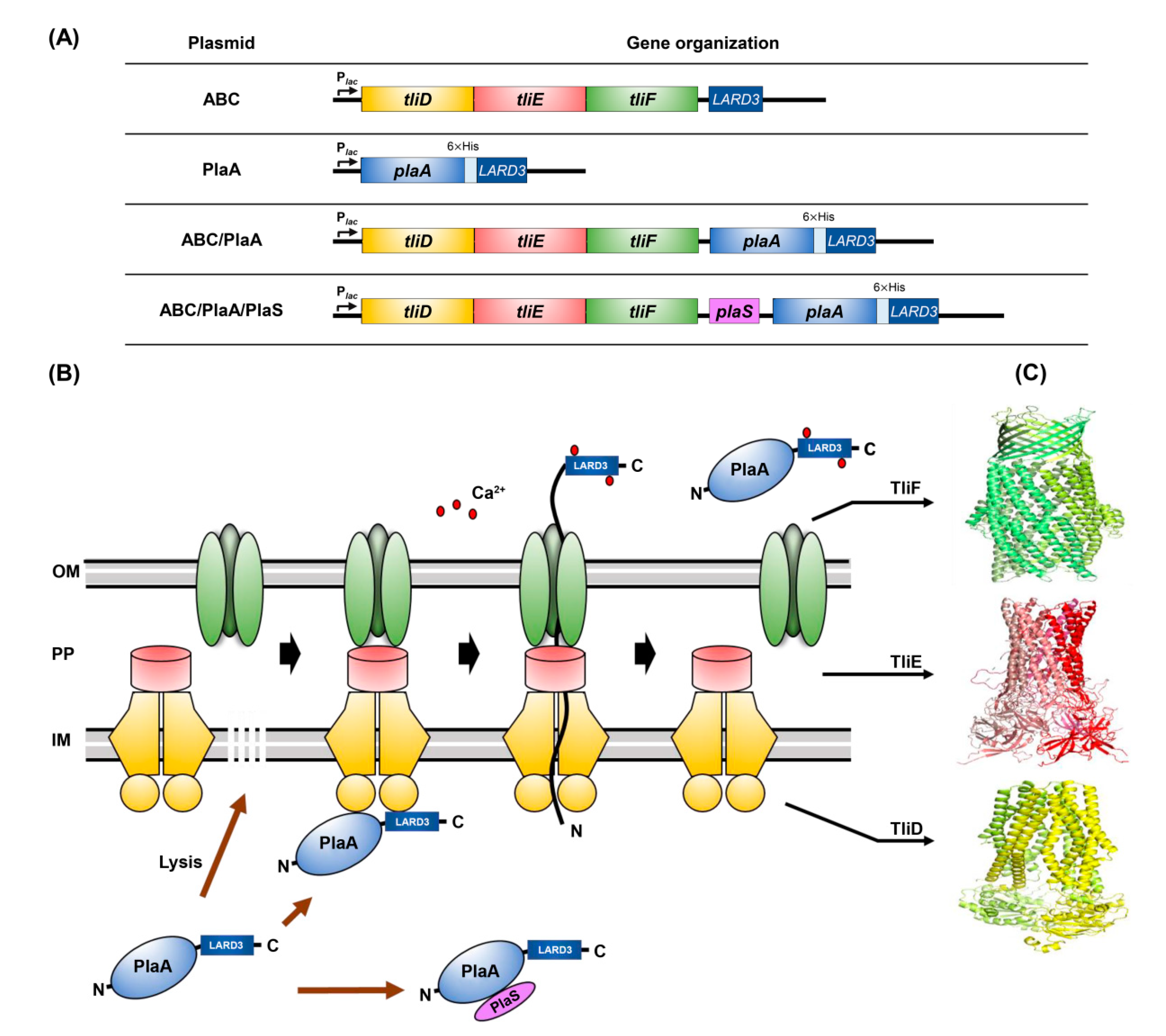



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer ID | Primer Sequence for PCR | Target Vector |
|---|---|---|
| lacI | F 5’ GCGGGGTTTTTTTTTAAGGCAGTTATTGGTCCCT 3’ R 5’ AAAAAGCCGCCAGCGGAACTGGCGGCGTGTGAAATTGTTATCCGCTC 3’ | pK19 |
| algD1 | F 5’ GACGGCCAGTGAATTCCACGAAGTGGTCGGCGTAGA3’ R 5’ AAAAAAAAACCCCGCCGAAGCGGGGTCAGTCGACGCCGGCTTTCTTGC3’ | pK19 |
| algD2 | F 5’ CGCTGGCGGCTTTTTTCCCAGTACTACATGCGCCC3’ R 5’ TGATTACGCCAAGCTGTTGAGCAGGGACGACACGT3’ | pK19 |
| lacZYA | F 5’ GGG AAGCTTGCGCAACGCAATTAATGTGAG 3’ R 5’ GGGCTGCAGGGTCAAAGAGGCATGATGCG 3’ | pK-lacI |
| plaA-1 | F 5’ TCTAGA ATGAGTATGTCTTTGAGTTTTAC 3’ R 5’ GGTACCGTGATGGTGATGGTGATGGGCATTGGCCATCGCCTCC 3’ | pDART |
| plaS-1 | F 5’ CAAGACAATGTCTAGGCCATGGGAGGCGATGGCC 3’ R 5’ ACATACTCATTCTAGCTCCTTGTCGTTACTGCTGTCCGTATTGCG 3’ | pDART |
| plaA-2 | F 5’ GCATGCCTAGCGACAAGGAGTCGGCATGA 3’ R 5 ’TCTAGATTAGTGATGGTGATGGTGATGGGCATTG GCCATCGCCTC 3’ | pDSK519 |
| plaS-2 | F 5’ TCTAGAGCCATGGGAGGCGATGGC 3’ R 5’ AGCTCTTACTGCTGTCCGTATTGCG 3’ | pDSK519 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Tae Eom, G.; Young Oh, J.; Hyun Park, J.; Chang Kim, S.; Kwang Song, J.; Hoon Ahn, J. High-Level Production of Bacteriotoxic Phospholipase A1 in Bacterial Host Pseudomonas fluorescens via ABC Transporter-Mediated Secretion and Inducible Expression. Microorganisms 2020, 8, 239. https://doi.org/10.3390/microorganisms8020239
Park J, Tae Eom G, Young Oh J, Hyun Park J, Chang Kim S, Kwang Song J, Hoon Ahn J. High-Level Production of Bacteriotoxic Phospholipase A1 in Bacterial Host Pseudomonas fluorescens via ABC Transporter-Mediated Secretion and Inducible Expression. Microorganisms. 2020; 8(2):239. https://doi.org/10.3390/microorganisms8020239
Chicago/Turabian StylePark, Jiyeon, Gyeong Tae Eom, Joon Young Oh, Ji Hyun Park, Sun Chang Kim, Jae Kwang Song, and Jung Hoon Ahn. 2020. "High-Level Production of Bacteriotoxic Phospholipase A1 in Bacterial Host Pseudomonas fluorescens via ABC Transporter-Mediated Secretion and Inducible Expression" Microorganisms 8, no. 2: 239. https://doi.org/10.3390/microorganisms8020239