The Uropathogenic Specific Protein Gene usp from Escherichia coli and Salmonella bongori is a Novel Member of the TyrR and H-NS Regulons

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Plasmids
2.2. Computational Analysis and Genomic Island Sequence Comparisons
2.3. Recombinant DNA Manipulations
2.4. Minimal Inhibitory Concentrations (MICs) for Chloramphenicol
2.5. β-Galactosidase Assays
2.6. Western Immunoblotting
2.7. Protein Isolation and Electrophoretic Mobility Shift Assay (EMSA)
2.8. Statistical Analysis
3. Results
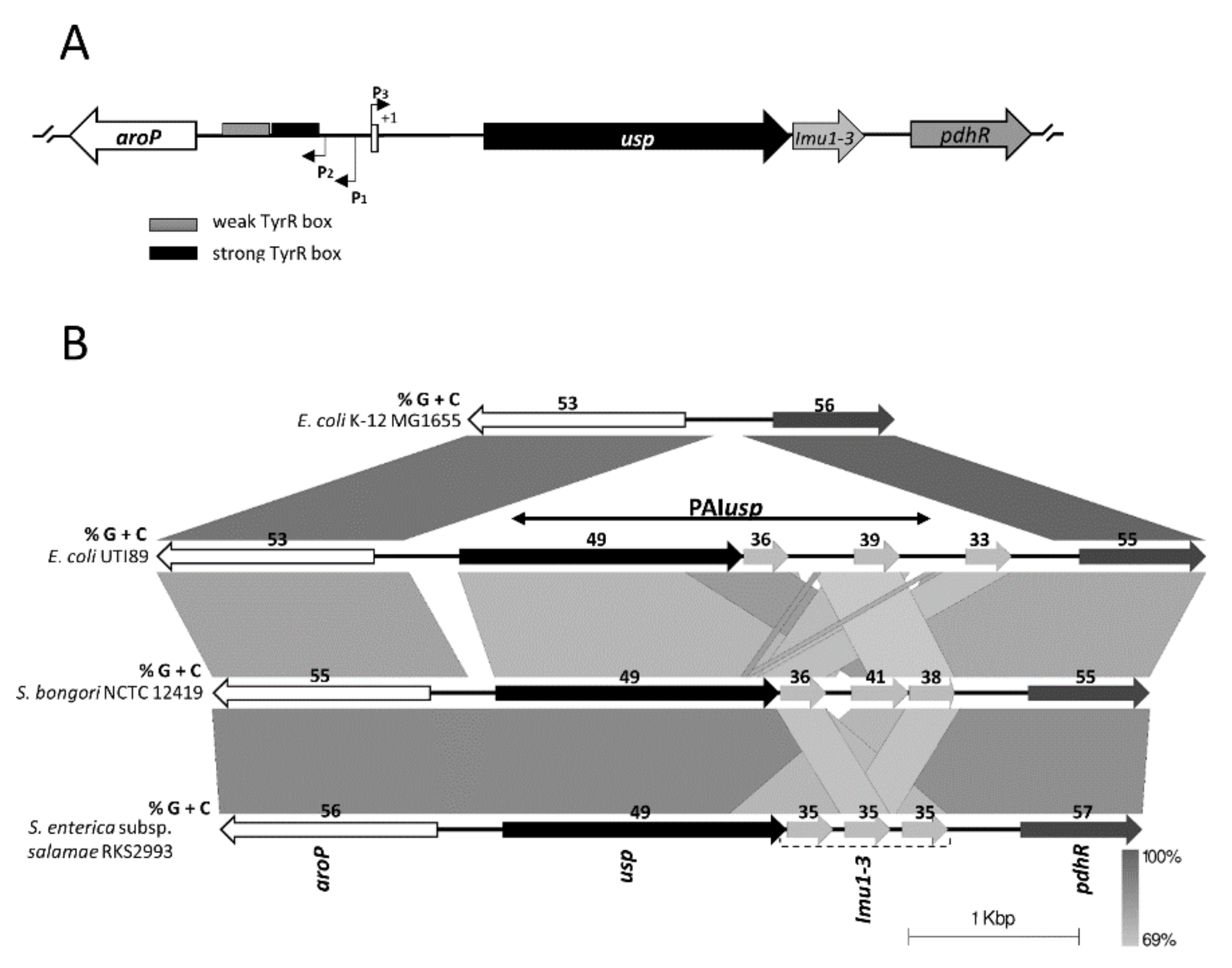
3.1. Bioinformatic Analysis of PAIusp prevalence
3.2. Expression of usp from the Divergent aroP P3 Promoter
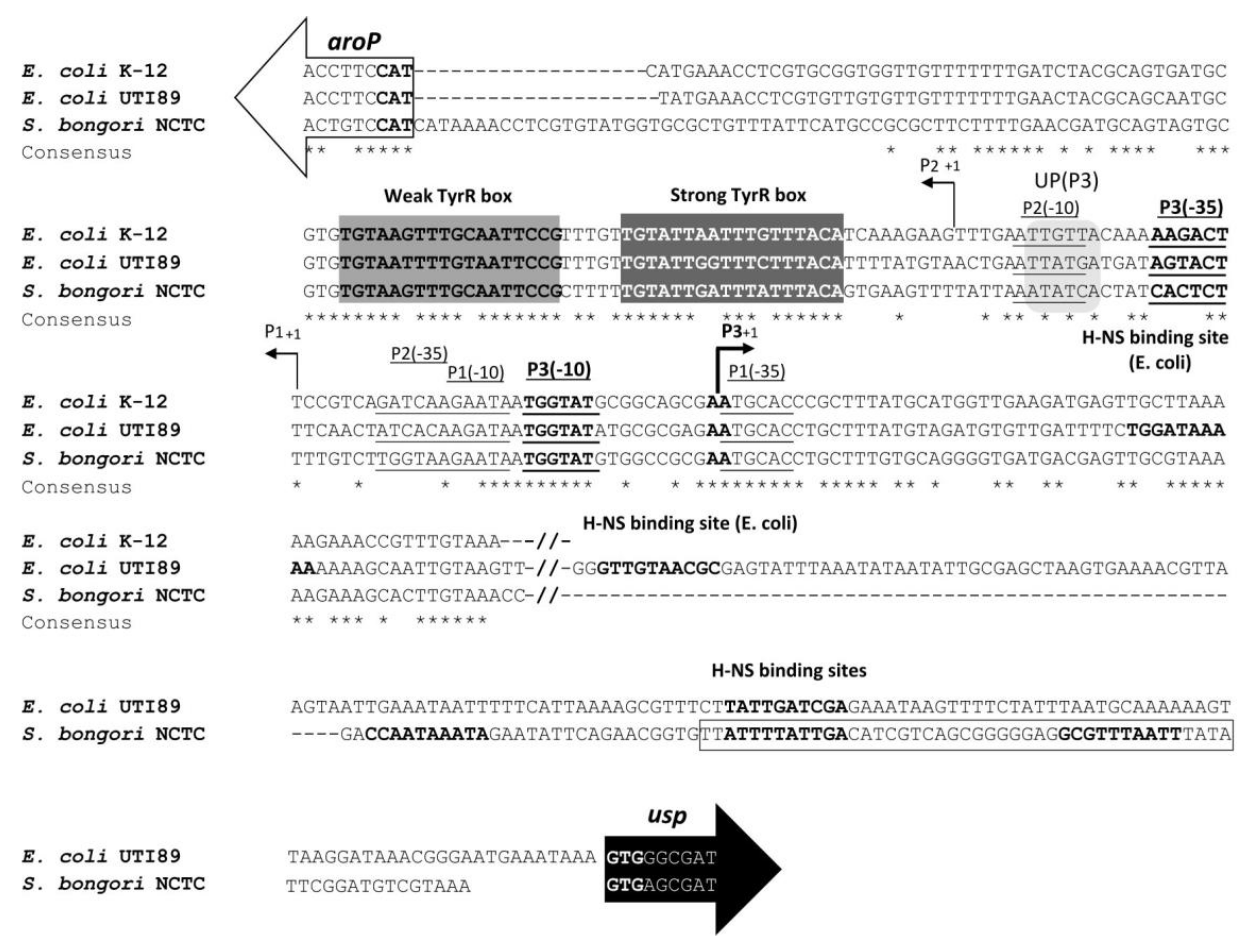
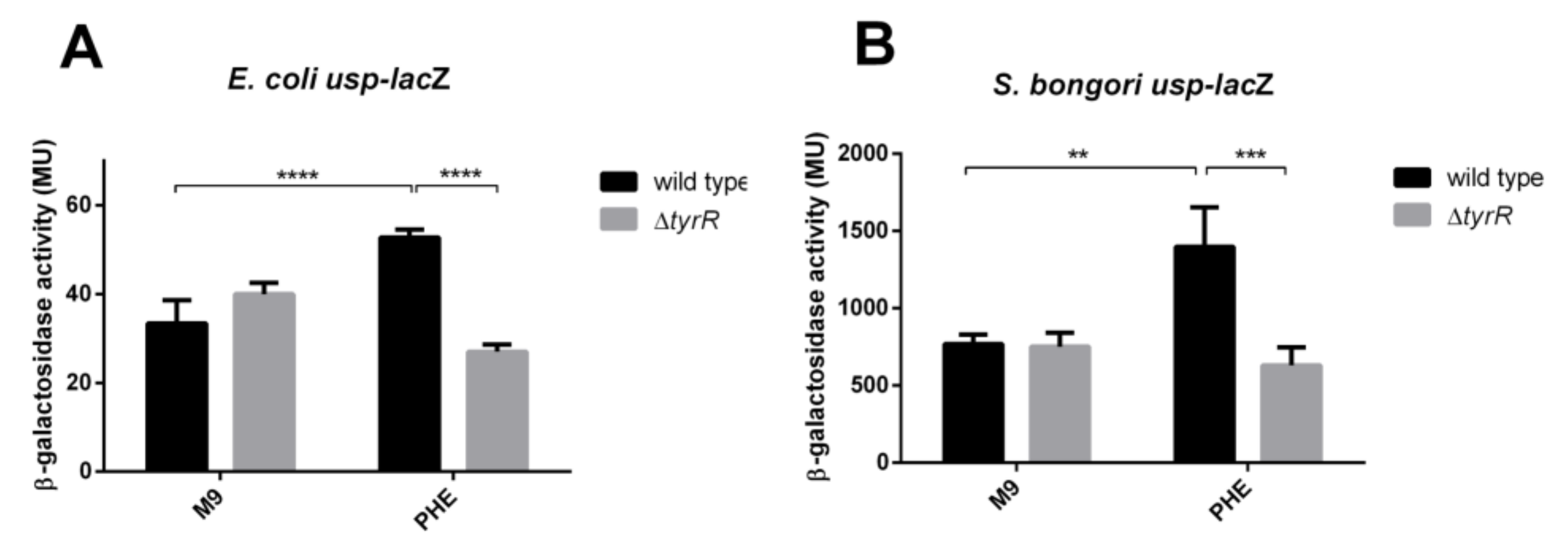
3.3. Positive Regulation of the usp P3 Promoter
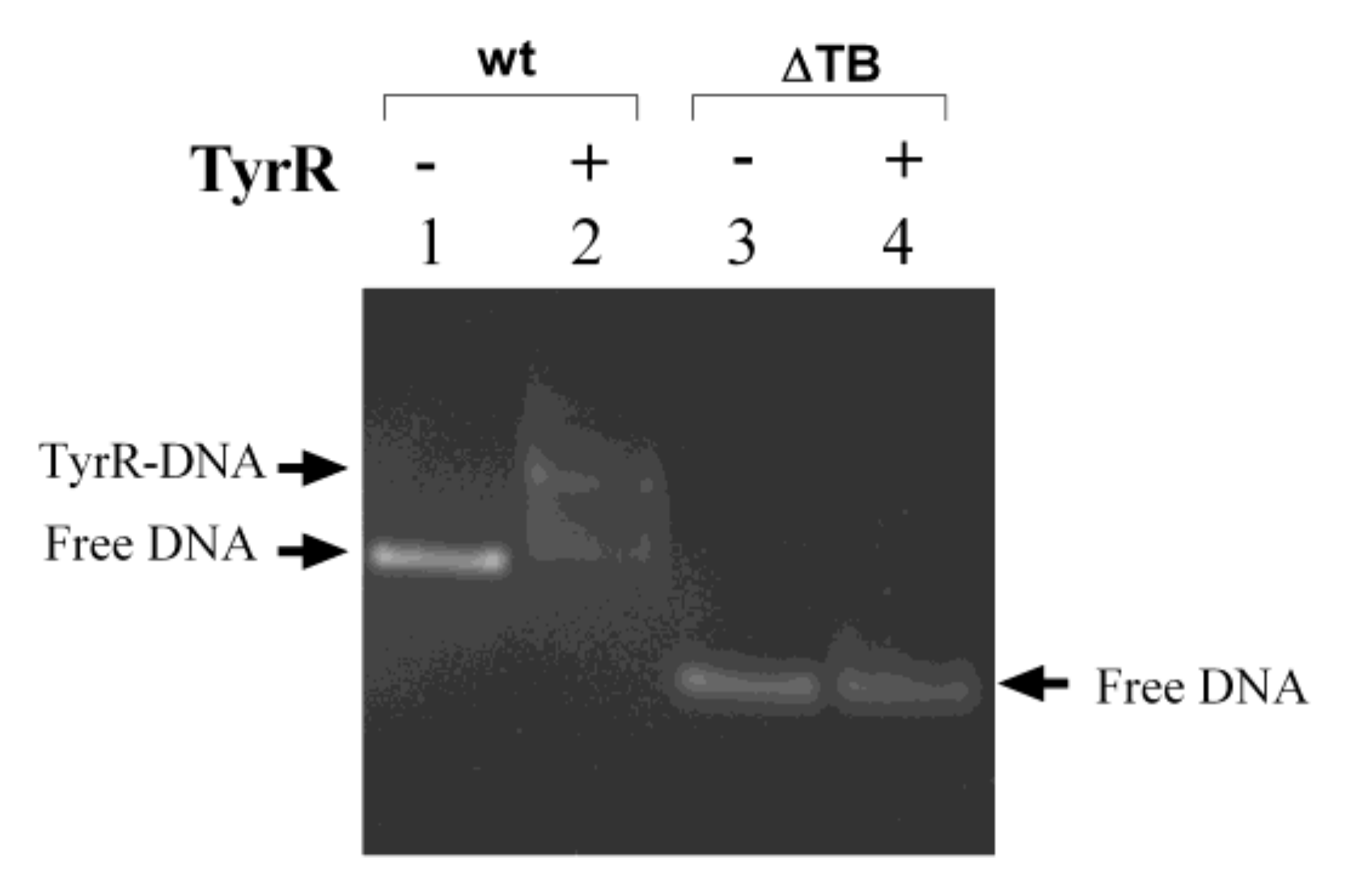
3.4. TyrR Binding to the S. bongori aroP-usp TyrR Boxes.
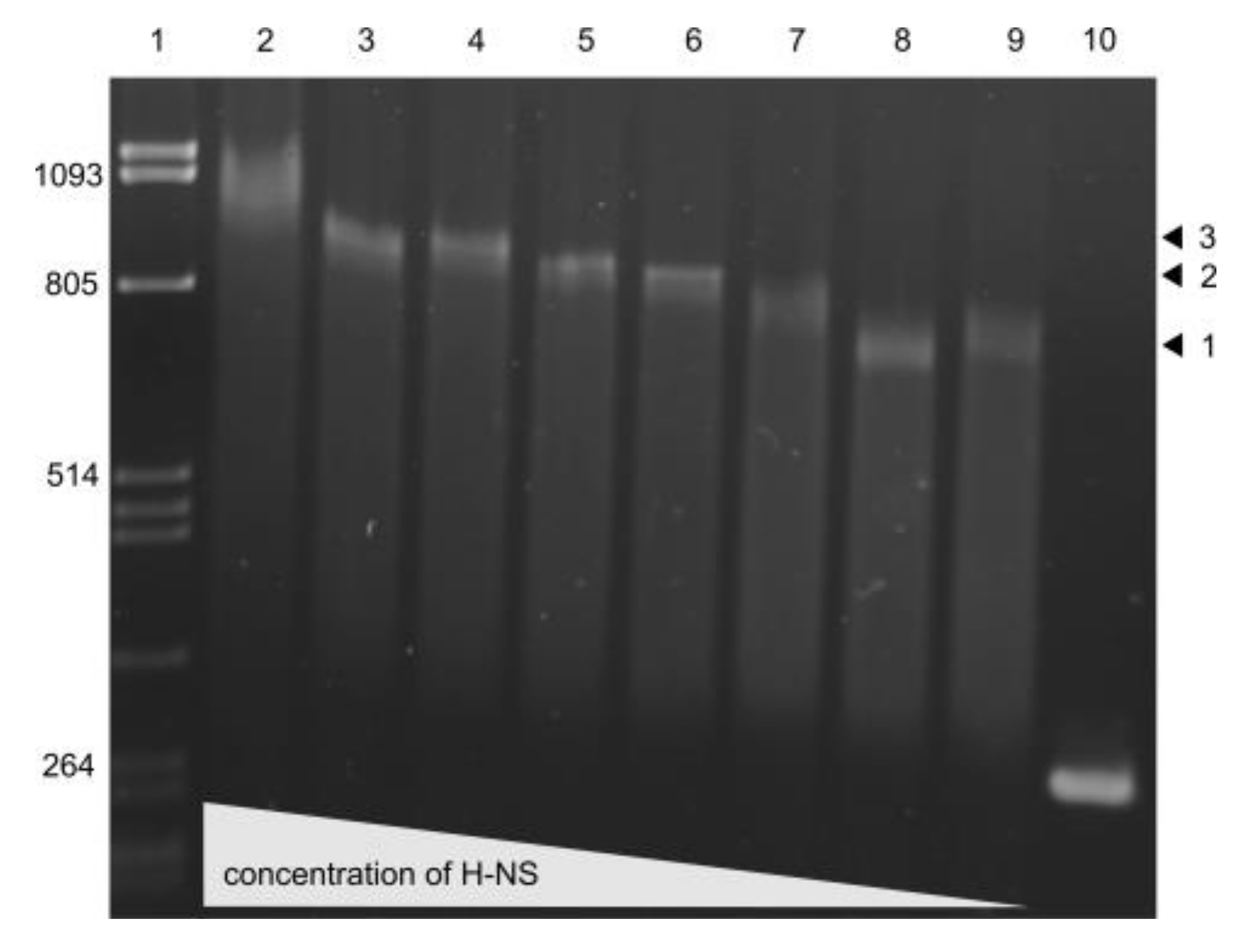
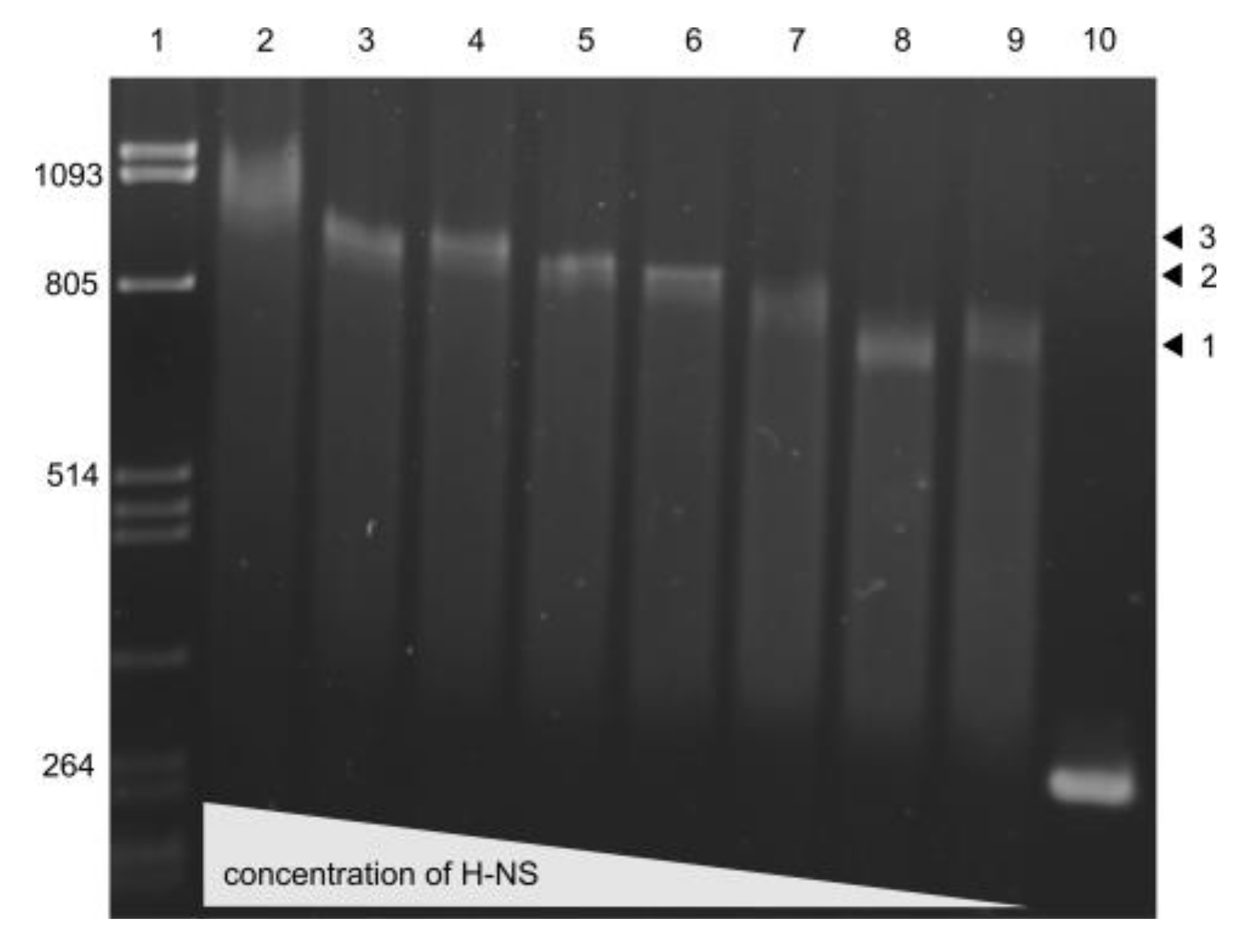
3.5. Repression of E. coli and S. bongori usp Expression by H-NS
3.6. H-NS binding to usp Promoter Region
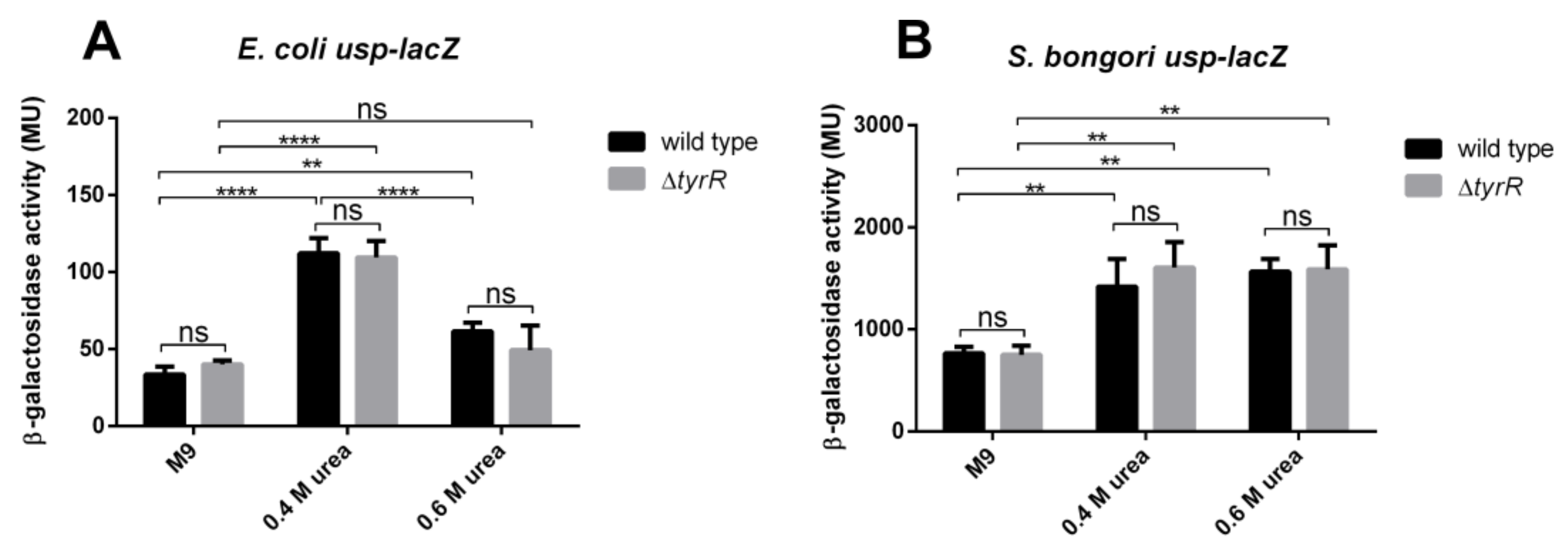
3.7. Urea Mediated Activation of Promoter P3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Crosa, J.H.; Brenner, D.J.; Ewing, W.H.; Falkow, S. Molecular relationships among the Salmonelleae. J. Bacteriol. 1973, 115, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doolittle, R.F.; Feng, D.F.; Tsang, S.; Cho, G.; Little, E. Determining divergence times of the major kingdoms of living organisms with a protein clock. Science 1996, 271, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuiston, J.R.; Fields, P.I.; Tauxe, R.V.; Logsdon, J.M.J. Do Salmonella carry spare tyres? Trends Microbiol. 2008, 16, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, A.; Mammina, C.; Villafrate, M.R.; Massenti, M.F.; Scarlata, G.; Diquattro, M. Multiple typing of strains of Salmonella enterica subsp. bongori ser. 48:Z35: Isolated in southern Italy. Ann. Inst. Pasteur Microbiol. 1988, 139, 605–612. [Google Scholar] [CrossRef]
- Giammanco, G.M.; Pignato, S.; Mammina, C.; Grimont, F.; Grimont, P.A.; Nastasi, A. Persistent endemicity of Salmonella bongori 48: Z (35): In Southern Italy: Molecular characterization of human, animal, and environmental isolates. J. Clin. Microbiol. 2002, 40, 3502–3505. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.J.A.; Cernela, N.; Muller, A.; Stephan, R.; Bloemberg, G. Draft Genome Sequence of Salmonella bongori N19-781, a Clinical Strain from a Patient with Diarrhea. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Abbott, S.L.; Ni, F.C.Y.; Janda, J.M. Increase in extraintestinal infections caused by Salmonella enterica subspecies II–IV. Emerg. Infect. Dis. 2012, 18, 637–639. [Google Scholar] [CrossRef]
- Nair, S.; Wain, J.; Connell, S.; de Pinna, E.; Peters, T. Salmonella enterica subspecies II infections in England and Wales—The use of multilocus sequence typing to assist serovar identification. J. Med. Microbiol. 2014, 63, 831–834. [Google Scholar] [CrossRef]
- Blum, G.; Ott, M.; Lischewski, A.; Ritter, A.; Imrich, H.; Tschäpe, H.; Hacker, J. Excision of large DNA regions termed pathogenicity islands from tRNA-specific loci in the chromosome of an Escherichia coli wild-type pathogen. Infect. Immun. 1994, 62, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Seth-Smith, H.M.B.; Fookes, M.C.; Okoro, C.K.; Baker, S.; Harris, S.R.; Scott, P.; Pickard, D.; Quail, M.A.; Churcher, C.; Sanders, M.; et al. Structure, diversity, and mobility of the Salmonella pathogenicity island 7 family of integrative and conjugative elements within Enterobacteriaceae. J. Bacteriol. 2012, 194, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Nakano, M.; Terai, A.; Yuri, K.; Nakata, K.; Nair, G.B.; Kurazono, H.; Ogawa, O. The presence of the virulence island containing the USP gene in uropathogenic Escherichia coli is associated with urinary tract infection in an experimental mouse model. J. Urol. 2001, 165, 1347–1351. [Google Scholar] [CrossRef]
- Lloyd, A.L.; Rasko, D.A.; Mobley, H.L. Defining genomic islands and uropathogen-specific genes in uropathogenic Escherichia coli. J. Bacteriol. 2007, 189, 3532–3546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nipič, D.; Podlesek, Z.; Budič, M.; Črnigoj, M.; Žgur-Bertok, D. Escherichia coli uropathogenic-specific protein, Usp, is a bacteriocin-like genotoxin. J. Infect. Dis. 2013, 208, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Grasso, F.; Frisan, T. Bacterial Genotoxins: Merging the DNA Damage Response into Infection Biology. Biomolecules 2015, 5, 1762–1782. [Google Scholar] [CrossRef] [Green Version]
- Nougayrede, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Nat. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [Green Version]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated Escherichia coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [Green Version]
- Roche-Lima, A.; Carrasquillo-Carrión, K.; Gómez-Moreno, R.; Cruz, J.M.; Velázquez-Morales, D.M.; Rogozin, I.B.; Baerga-Ortiz, A. The presence of genotoxic and/or pro-inflammatory bacterial genes in gut metagenomic databases and their possible link with inflammatory bowel diseases. Front. Genet. 2018, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yang, J.; Lawley, B.; Pittard, A.J. Repression of the aroP gene of Escherichia coli involves activation of a divergent promoter. J. Bacteriol. 1997, 179, 4213–4218. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yang, J.; Pittard, A.J. Promoters and transcripts associated with the aroP gene of Escherichia coli. J. Bacteriol. 1997, 179, 4206–4212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Yang, J.; Pittard, A.J. Demonstration that the TyrR protein and RNA polymerase complex formed at the divergent P3 promoter inhibits binding of RNA polymerase to the major promoter, P1, of the aroP gene of Escherichia coli. J. Bacteriol. 1998, 180, 5466–5472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittard, J.; Camakaris, H.; Yang, J. The TyrR regulon. Mol. Microbiol. 2005, 55, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Sarsero, J.P.; Pittard, A.J. Molecular analysis of the TyrR protein-mediated activation of mtr gene expression in Escherichia coli K-12. J. Bacteriol. 1991, 173, 7701–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawley, B.; Fujita, N.; Ishihama, A.; Pittard, A.J. The TyrR protein of Escherichia coli is a class I transcription activator. J. Bacteriol. 1995, 177, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainger, D.C. Structure and function of bacterial H-NS protein. Biochem. Soc. Trans. 2016, 44, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2, 2006.0008. [Google Scholar] [CrossRef] [Green Version]
- Lodge, J.; Fear, J.; Busby, S.; Gunasekaran, P.; Kamini, N.R. Broad host range plasmids carrying the Escherichia coli lactose and galactose operons. FEMS Microbiol. Lett. 1992, 74, 271–276. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Maccallum, P.; Russel, D. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbour Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, S.F. Mutagenesis by incorporation of a phosphorylated oligo during PCR amplification. Biotechniques 1994, 16, 410–412. [Google Scholar] [PubMed]
- Miller, J.H. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1974. [Google Scholar]
- Molan, K.; Podlesek, Z.; Hodnik, V.; Butala, M.; Oswald, E.; Žgur Bertok, D. The Escherichia coli colibactin resistance protein ClbS is a novel DNA binding protein that protects DNA from nucleolytic degradation. DNA Repair. 2019, 79, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Marti, R.; Hagens, S.; Loessner, M.J.; Klumpp, J. Genome Sequence of Salmonella bongori Strain N268-08. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Parret, A.H.; De Mot, R. Escherichia coli’s uropathogenic-specific protein: A bacteriocin promoting infectivity? Microbiology 2002, 148, 1604–1606. [Google Scholar] [CrossRef]
- Harley, C.B.; Reynolds, R.P. Analysis of Escherichia coli promoter sequences. Nucleic Acids Res. 1987, 15, 2343–2361. [Google Scholar] [CrossRef]
- Chen, H.; Tang, H.; Ebright, R.H. Functional interaction between RNA polymerase α subunit C-terminal domain and σ70 in UP-element and activator-dependent transcription. Mol. Cell 2003, 11, 1621–1633. [Google Scholar] [CrossRef]
- Estrem, S.T.; Ross, W.; Gaal, T.; Chen, Z.W.S.; Niu, W.; Ebright, R.H.; Gourse, R.L. Bacterial promoter architecture: Subsite structure of UP elements and interactions with the carboxyterminal domain of the RNA polymerase α subunit. Genes Dev. 1999, 13, 2134–2147. [Google Scholar] [CrossRef]
- Münch, R.; Hiller, K.; Grote, A.; Scheer, M.; Klein, J.; Schobert, M.; Jahn, D. VirtualFootprint and PRODORIC: An integrative framework for regulon prediction in prokaryotes. Bioinformatics 2005, 21, 4187–4189. [Google Scholar] [CrossRef] [Green Version]
- Culham, D.E.; Lu, A.; Jishage, M.; Krogfelt, K.A.; Ishihama, A.; Wood, J.M. The osmotic stress response and virulence in pyelonephritis isolates of Escherichia coli: Contributions of RpoS, ProP, ProU and other systems. Microbiology 2001, 147, 1657–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarre, W.W. The impact of gene silencing on horizontal gene transfer and bacterial evolution. Adv. Microb. Physiol. 2016, 69, 157–186. [Google Scholar] [PubMed]
- Hayek, N. Lateral transfer and GC content of bacterial resistance genes. Front. Microbiol. 2013, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Ravenhall, M.; Škunca, N.; Lassalle, F.; Dessimoz, C. Inferring Horizontal Gene Transfer. PLoS Comput. Biol. 2015, 11, e1004095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fookes, M.; Schroeder, G.N.; Langridge, G.C.; Blondel, C.J.; Mammina, C.; Connor, T.R.; Seth-Smith, H.; Vernikos, G.S.; Robinson, K.S.; Sanders, M.; et al. Salmonella bongori provides insights into the evolution of the Salmonellae. PLoS Pathog. 2011, 7, e1002191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Gorman, L.E.; Krejany, E.O.; Bennett-Wood, V.R.; Robins-Browne, R.M. Transfer of attaching and effacing capacity from a strain of enteropathogenic Escherichia coli to E. coli K-12. Microbiol. Res. 1996, 151, 379–385. [Google Scholar] [CrossRef]
- Elliott, S.J.; Yu, J.; Kaper, J.B. The cloned locus of enterocyte effacement from enterohemorrhagic Escherichia coli O157:H7 is unable to confer the attaching and effacing phenotype upon E. coli K-12. Infect. Immun. 1999, 67, 4260–4263. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.; Petty, N.K.; Hocking, D.; Bennett-Wood, V.; Wakefield, M.; Praszkier, J.; Tauschek, M.; Yang, J.; Robins-Browne, R. Evolutionary adaptation of an AraC-like regulatory protein in Citrobacter rodentium and Escherichia species. Infect. Immun. 2015, 83, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Liu, Z.; He, J.; Wang, J.; Yan, Y.; Wang, X.; Cui, Y.; Yujing, B.; Zongmin, D.; Yajun, S.; et al. TyrR, the regulator of aromatic amino acid metabolism, is required for mice infection of Yersinia pestis. Front. Microbiol. 2015, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- Palace, S.G.; Proulx, M.K.; Lu, S.; Baker, R.E.; Goguen, J.D. Genome-wide mutant fitness profiling identifies nutritional requirements for optimal growth of Yersinia pestis in deep tissue. MBio 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Cersini, A.; Salvia, A.M.; Bernardini, M.L. Intracellular multiplication and virulence of Shigella flexneri auxotrophic mutants. Infect. Immun. 1998, 66, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stritzker, J.; Janda, J.; Schoen, C.; Taupp, M.; Pilgrim, S.; Gentschev, I.; Schreier, P.; Geginat, G.; Goebel, W. Growth, virulence, and immunogenicity of Listeria monocytogenesaro mutants. Infect. Immun. 2004, 72, 5622–5629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shindo, H.; Ohnuki, A.; Ginba, H.; Katoh, E.; Ueguchi, C.; Mizuno, T.; Yamazaki, T. Identification of the DNA binding surface of H-NS protein from Escherichia coli by heteronuclear NMR spectroscopy. FEBS Lett. 1999, 455, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Gordon, B.R.; Li, Y.; Cote, A.; Weirauch, M.T.; Ding, P.; Hughes, T.R.; Navarre, W.W.; Xia, B.; Liu, J. Structural basis forrecognition of AT-rich DNA by unrelated xenogeneic silencing proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 10690–10695. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Foo, Y.H.; Winardhi, R.S.; Tang, Q.; Yan, J.; Kenney, L.J. Charged residues in the H-NS linker drive DNA binding and gene silencing in single cells. Proc. Natl. Acad. Sci. USA 2017, 114, 12560–12565. [Google Scholar] [CrossRef] [Green Version]
- Ono, S.; Goldberg, M.D.; Olsson, T.; Esposito, D.; Hinton, J.C.; Ladbury, J.E. H-NS is a part of a thermally controlled mechanism for bacterial gene regulation. Biochem. J. 2005, 391, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Stella, S.; Falconi, M.; Lammi, M.; Gualerzi, C.O.; Pon, C.L. Environmental control of the in vivo oligomerization of nucleoid protein H-NS. J. Mol. Biol. 2006, 355, 169–174. [Google Scholar] [CrossRef]
- Prosseda, G.; Falconi, M.; Giangrossi, M.; Gualerzi, C.O.; Micheli, G.; Colonna, B. The virF promoter in Shigella: More than just a curved DNA stretch. Mol. Microbiol. 2004, 51, 523–537. [Google Scholar] [CrossRef]
- Shahul Hameed, U.F.; Liao, C.; Radhakrishnan, A.K.; Huser, F.; Aljedani, S.S.; Zhao, X.; Momin, A.A.; Melo, F.A.; Guo, X.; Brooks, C.; et al. H-NS uses an autoinhibitory conformational switch for environment-controlled gene silencing. Nucleic Acids Res. 2019, 47, 2666–2680. [Google Scholar] [CrossRef] [Green Version]
- Zeidel, M.L.; Ambudkar, S.V.; Smith, B.L.; Agre, P. Reconstitution of functional water channels in liposomes containing purified red cell CHIP28 protein. Biochemistry 1992, 31, 7436–7440. [Google Scholar] [CrossRef]
- Withman, B.; Gunasekera, T.S.; Beesetty, P.; Agans, R.; Paliy, O. Transcriptional responses of uropathogenic Escherichia coli to increased environmental osmolarity caused by salt or urea. Infect. Immun. 2013, 81, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
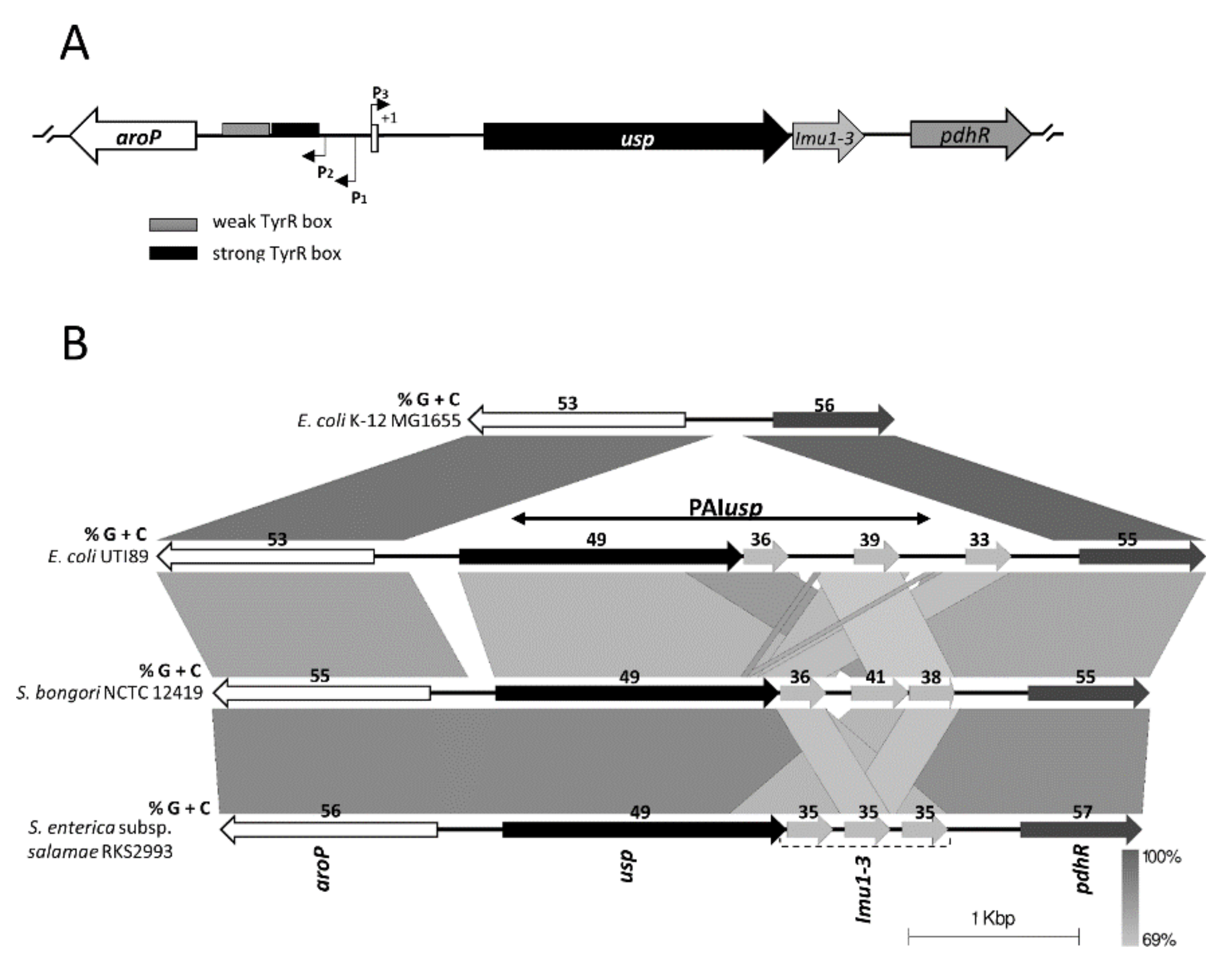
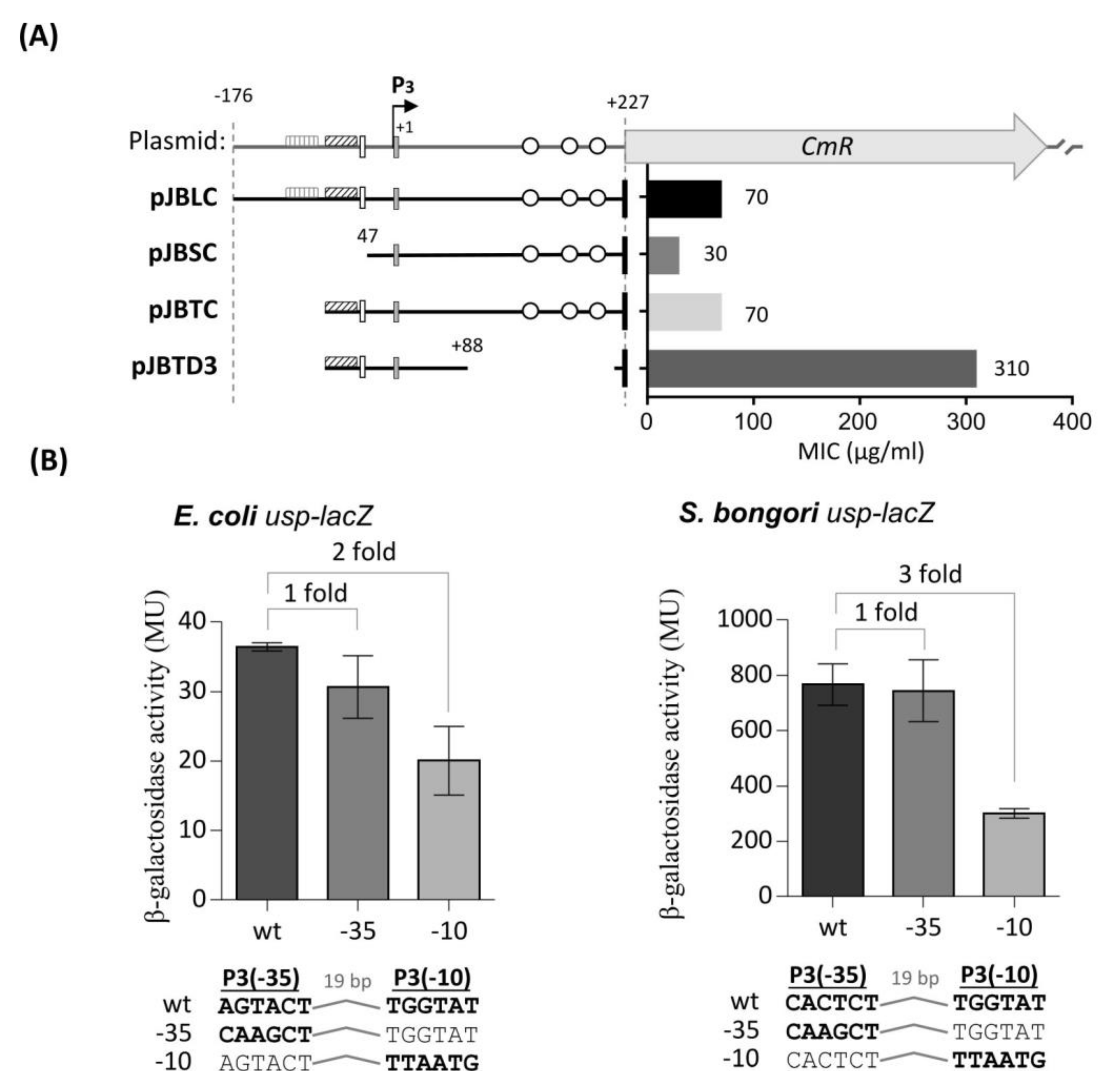
 ) and weak (
) and weak (  ) TyrR boxes. The UP element is designated as an open square and H-NS binding sites as open circles. The ability of the fragments to drive CAT expression in usp-CAT transcriptional fusions was determined indirectly by measuring MIC of chloramphenicol. (B) Effects of site-specific mutations in either the -35 or -10 site of P3. Units for β-galactosidase assay are those defined by Miller, 1974 [35]. The values of β-galactosidase activities are averages of three independent experiments, with standard deviations below 15%. Values above columns are fold repression: specific activity of β-galactosidase of the wild-type promoters divided by that of site-specific mutated promoters.
) and weak ( ) TyrR boxes. The UP element is designated as an open square and H-NS binding sites as open circles. The ability of the fragments to drive CAT expression in usp-CAT transcriptional fusions was determined indirectly by measuring MIC of chloramphenicol. (B) Effects of site-specific mutations in either the -35 or -10 site of P3. Units for β-galactosidase assay are those defined by Miller, 1974 [35]. The values of β-galactosidase activities are averages of three independent experiments, with standard deviations below 15%. Values above columns are fold repression: specific activity of β-galactosidase of the wild-type promoters divided by that of site-specific mutated promoters.
) TyrR boxes. The UP element is designated as an open square and H-NS binding sites as open circles. The ability of the fragments to drive CAT expression in usp-CAT transcriptional fusions was determined indirectly by measuring MIC of chloramphenicol. (B) Effects of site-specific mutations in either the -35 or -10 site of P3. Units for β-galactosidase assay are those defined by Miller, 1974 [35]. The values of β-galactosidase activities are averages of three independent experiments, with standard deviations below 15%. Values above columns are fold repression: specific activity of β-galactosidase of the wild-type promoters divided by that of site-specific mutated promoters.
) and weak ( ) TyrR boxes. The UP element is designated as an open square and H-NS binding sites as open circles. The ability of the fragments to drive CAT expression in usp-CAT transcriptional fusions was determined indirectly by measuring MIC of chloramphenicol. (B) Effects of site-specific mutations in either the -35 or -10 site of P3. Units for β-galactosidase assay are those defined by Miller, 1974 [35]. The values of β-galactosidase activities are averages of three independent experiments, with standard deviations below 15%. Values above columns are fold repression: specific activity of β-galactosidase of the wild-type promoters divided by that of site-specific mutated promoters.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains or Plasmids | Relevant Charasteristic(s) # | Reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | Cloning host; F– Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK−, mK+) phoA supE44 λ– thi-1 gyrA96 relA1 | Life Technologies |
| E. coli BL21 (DE3) | F−ompT hsd SB(rB−mB−) dcmgalλ (DE3) | Invitrogen |
| E. coli JW1889 | F-Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ−, ΔaraF751::kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 | [27] |
| E. coli JW1316 E. coli JW1225 | F-Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ−, ΔtyrR760::kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 F-Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ−, Δhns746:: kan, rph-1, Δ(rhaD-rhaB)568, hsdR514 | [27] |
| E. coli TA211 | wild-type strain (UPEC isolate) | IMI |
| E. coli MG1655 | wild-type strain | This laboratory |
| S.bongori NCTC 12419 | wild-type strain | ATTC |
| Plasmids | ||
| pRW50 | TcR; Low-copy-number promoterless lacZ trancriptional fusion vector | [28] |
| pRWBP | TcR; 403 bp EcoRI-HindIII fragment from S. bongori NCTC 12419 aroP-usp intergenic region: usp-lacZ transcription fusion | This study |
| pRWBPM1 | TcR; pRWBP derivate; P3 (−35) mutation: CACTCT-->CAAGCT | This study |
| pRWBPM3 | TcR; pRWBP derivate; P3 (−10) mutation: TGGTAT-->TTAATG | This study |
| pRCPL | TcR; 527 bp EcoRI-HindIII fragment from E. coli TA211 aroP-usp intergenic region: usp-lacZ trancription fusion | This study |
| pRWC10 | TcR; pRCPL derivate; P3 (−10) mutation: TGGTAT-->TTAATG | This study |
| pRWC35 | TcR; pRCPL derivate; P3 (−10) mutation: TGGTAT-->TTAATG | This study |
| pRWMGA- | TcR; 215 bp EcoRI-HindIII fragment from E. coli K-12 MG1655aroP-pdhR intergenic region: aroP-lacZ transcription fusion | |
| pJCAT | ApR; High-copy-number promoterless cat trancription fusion vector (pJET1.2 cloning vector (Thermo Fisher Scientific) derivate) | This study |
| pJBLC | ApR; 403 bp PstI-BstEII fragment from S. bongori aroP-usp intergenic region: usp-cat transcription fusion | This study |
| pJBSC | ApR; pJBLC derivate; 247 bp fragment without both TyrR binging boxes | This study |
| pJBTC | ApR; pJBLC derivate; 315 bp fragment without weak TyrR binding box | This study |
| pJBTD2 | ApR; 111 bp PstI-BstEII fragment from S. bongori aroP-usp intergenic region only with strong TyrR box and P3 promoter | This study |
| pJBTD3 | ApR; pJBTD2 derivate: 111 bp fragment extended for 64 bp | This study |
| pET8HT | ApR; pET8c -derived expression vector with PT7 promoter: expression and production of recombinant N-terminally His6-tagged TyrR | This study |
| pET9HT | ApR; pET8c -derived expression vector with PT7 promoter: expression and production of recombinant N-terminally His6-tagged H-NS | This study |
| pUSP4R-L | ApR; T7 promoter of pUSP4 replaced with native E. coli TA211 usp promoter. | [14] |
| pCUPBI | ApR; T7 promoter of pUSP4 replaced with native S. bongori NCTC 12419 promoter | This study |
| Primer | Sequence (5′→3′) | Purpose |
|---|---|---|
| Cm_Bst_F | TTGGTAACCAAGAGAAAAAAATCACTGGATATAC | PCR amplification of cat gene from pLysS plasmid for cloning in pJBPLN3 (intermediate plasmid) |
| Cm_Xba_R | TTTCTAGATTACGCCCCGCCCTGCCACTCAT | |
| Bong_L_Pst_F | CTGCAGCATAAAACCTCGTGTATGGTGCGC | PCR amplification of S. bongori aroP-usp promoter region for cloning in pJBLC |
| Bong_S_Pst_F | CTGCAGCACTATCACTCTTTTGTCTTGGTAAG | PCR amplification of S. bongori aroP-usp promoter region for cloning in pJBSC and also used in EMSA |
| Bong_L_Bst_R | GGTTACCCATTTTACGACATCCGAATATAAATTAAACGCC | PCR amplification of S. bongori aroP-usp promoter region for cloning in pJBLC, pJBSCm, pJBTCm |
| Bong_promS_tyr_F | TTCTGCAGCTTTTGTATTGATTTATTTACAGTGA | PCR amplification of S. bongori aroP-usp promoter region for cloning in pJBTC, pJBTD2 and pJBTD3 |
| Bong_prom_del2_R | TTGGTTACCCATTTTACGACATCCGAACTCGTCATCACCCCTGCACAAAGCAGGTGCAT | PCR amplification of S. bongori aroP-usp promoter region for cloning in pJBTD2 |
| Bong_prom_del3_R | TTGGTTACCCATTTTACGACATCCGAATGCAAAATAAAGTGAGTAATGCTAAAAGGG | PCR amplification of S. bongori aroP-usp promoter region for cloning in pJBTD3 |
| Bong_L_Eco_F | GAATTCATAAAACCTCGTGTATGGTGCGC | PCR amplification of S. bongori aroP-usp promoter region for cloning in pRWBP, pRWBPM1, pRWBPM2 and also used in EMSA |
| Mut10_Vsp | TGTCTTGGTAAGAATAATTAATTGTGACTGCGAATGCAC | PCR amplification of S. bongori aroP-usp promoter region for cloning in pRWBP, pRWBPM1, pRWBPM2 |
| Mut35_Hind | ATTAAATATCACTATCAAGCTTTTGTCTTGGTAAGAA | PCR amplification of S. bongori aroP-usp promoter region for cloning in pRWBP, pRWBPM1, pRWBPM2 |
| Bong_prom_Hind_R | TTAAGCTTTACGACATCCGAATATAAATTAAACGCC | PCR amplification of S. bongori aroP-usp promoter region for cloning in pRWBP, pRWBPM1, pRWBPM2 and also used in EMSA |
| Coli_L_Eco_F | TTGAATTCTATGAAACCTCGTGTTGTGTTGTT | PCR amplification of E. coli 211 aroP-usp promoter region for cloning in pRCPL, pRWC10 and pRWC35 |
| Mut_coli_10_Vsp | ACTATCACAAGATAAATTAATATGCGCGAGAATGCACC | |
| Mut_coli_35_Hind | ACTGAATTATGATGATAAAGCTTTCAACTATCACAAG | |
| Coli_L _R_Hind | TTAAGCTTTATTTCATTCCCGTTTATCCTTAAC | |
| MG_prom_F_Eco | TTGAATTCGAAACCTCGTGCGGTGGTTGTTTTTTTG | PCR amplification of E. coli K-12 MG1655 aroP promoter region for cloning in pRWMGA- |
| MG_prom_R_Hind | TTAAGCTTTACAAACGGTTTCTTTTTAAGC | |
| TyrR_Bam_F | TTGGATCCCGTCTGGAAGTCTTTTGTGAAGACC | PCR amplification of tyrR gene from E. coli DH5α for cloning in pET8HT PCR amplification of hns gene from E. coli DH5α for cloning in pET9HT |
| TyrR_Mlu_R H-NS Bam F H-NS Mlu R | TTACGCGTTACTCTTCGTTCTTCTTCTGACTCAGAC TTGGATCCAGCGAAGCACTTAAAATTCTGAAC TTACGCGTTTATTGCTTGATCAGGAAATCGTCG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rihtar, E.; Žgur Bertok, D.; Podlesek, Z. The Uropathogenic Specific Protein Gene usp from Escherichia coli and Salmonella bongori is a Novel Member of the TyrR and H-NS Regulons. Microorganisms 2020, 8, 330. https://doi.org/10.3390/microorganisms8030330
Rihtar E, Žgur Bertok D, Podlesek Z. The Uropathogenic Specific Protein Gene usp from Escherichia coli and Salmonella bongori is a Novel Member of the TyrR and H-NS Regulons. Microorganisms. 2020; 8(3):330. https://doi.org/10.3390/microorganisms8030330
Chicago/Turabian StyleRihtar, Erik, Darja Žgur Bertok, and Zdravko Podlesek. 2020. "The Uropathogenic Specific Protein Gene usp from Escherichia coli and Salmonella bongori is a Novel Member of the TyrR and H-NS Regulons" Microorganisms 8, no. 3: 330. https://doi.org/10.3390/microorganisms8030330