Novel Clinical Campylobacter jejuni Infection Models Based on Sensitization of Mice to Lipooligosaccharide, a Major Bacterial Factor Triggering Innate Immune Responses in Human Campylobacteriosis


Abstract
:1. Introduction
2. Basic Concept and Aim of This Review Article
3. Human Campylobacteriosis
4. C. jejuni Lipooligosaccharide and Post-Infectious Sequelae in Human Campylobacteriosis
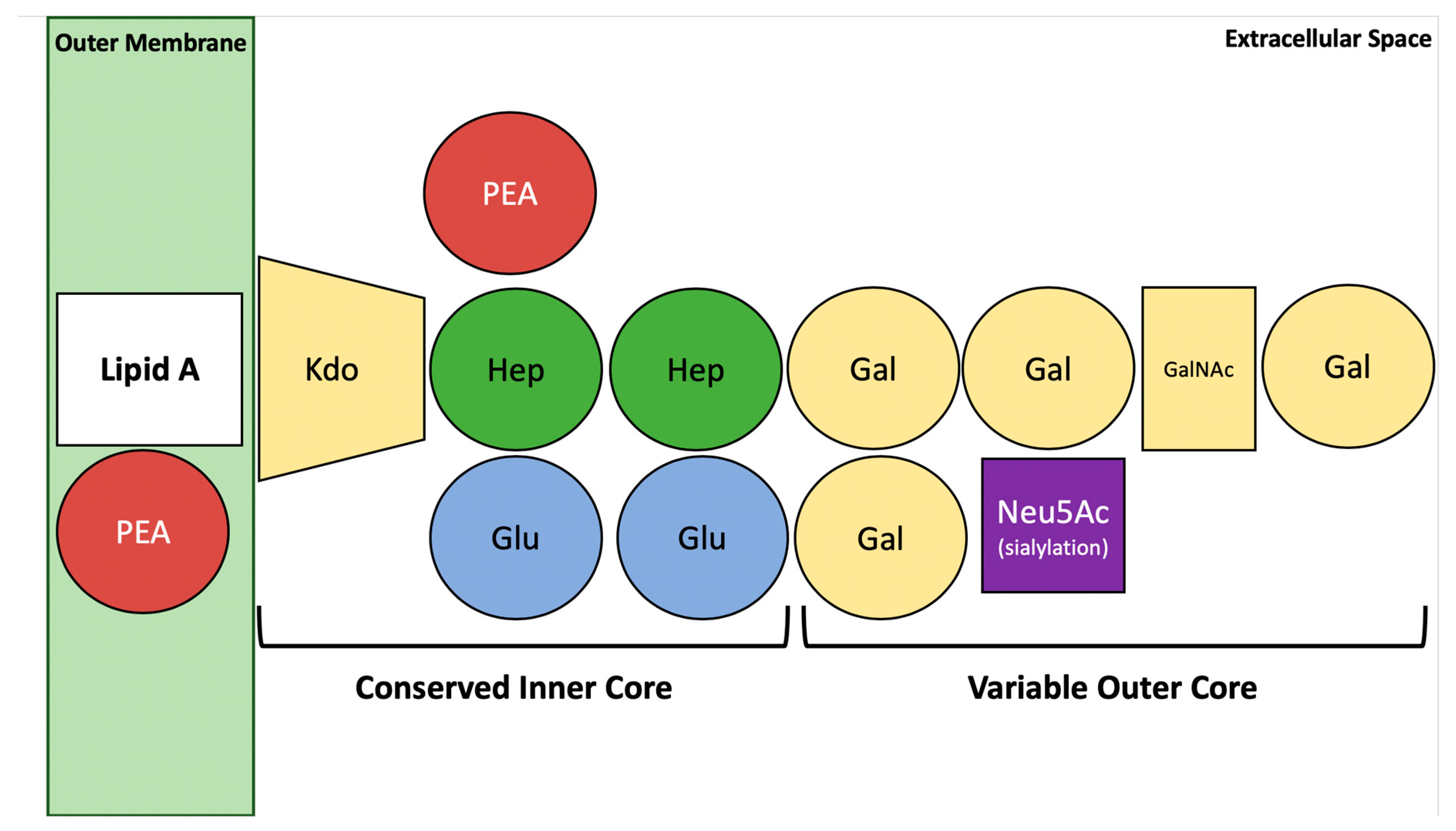
5. Structural Aspects of C. jejuni Lipooligosaccharide
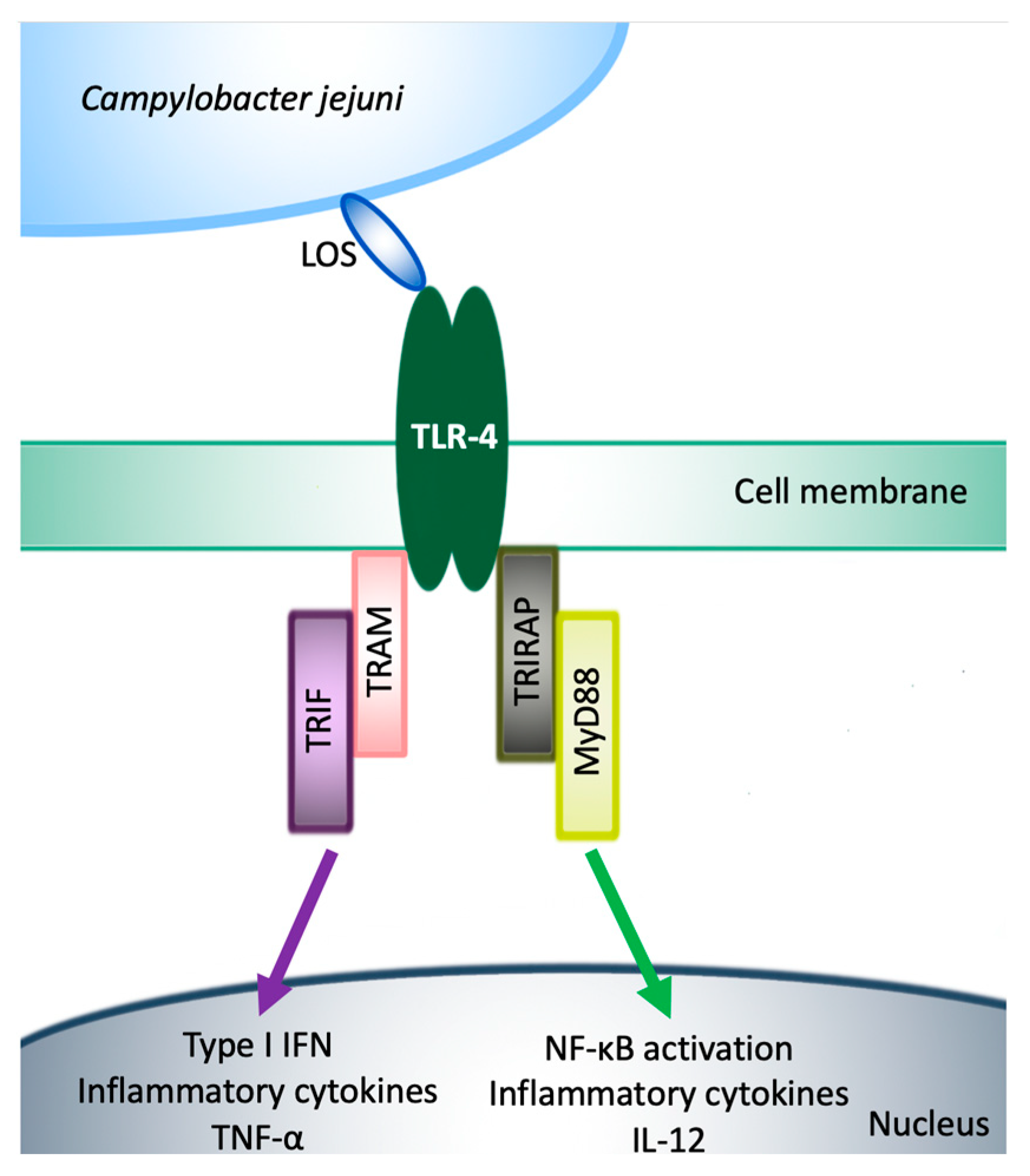
6. Activation of TLR-4 Signaling by C. jejuni Lipooligosaccharide in Humans and Mice
7. Generation of Secondary Abiotic Mice to Overcome Colonization Resistance
8. Rendering Mice Susceptible to LOS as Approach for Developing Valid C. jejuni Infection Models
9. The Use of Novel Murine Models of C. jejuni Infection in Actual Campylobacteriosis Research
10. Differences in LOS Tolerance among Humans and Birds Impede the Development of Prophylactic Measures against Campylobacteriosis
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CFU | colony forming unit |
| CPS | capsular polysaccharide |
| CR | colonization resistance |
| GBS | Guillain-Barré syndrome |
| IBD | inflammatory bowel disease |
| IBS | irritable bowel syndrome |
| IFN | interferon |
| Ig | immunoglobulin |
| IL | interleukin |
| Kdo | β-linked 3-deoxy-d-manno-oct-2-ulosonic acid |
| LOS | lipooligosaccharide |
| LPS | lipopolysaccharide |
| MCP-1 | monocyte chemoattractant protein 1 |
| MFS | Miller-Fisher syndrome |
| MIC | minimum inhibitory concentration |
| MLN | mesenteric lymph nodes |
| mRNA | messenger ribonucleic acid |
| mTOR | mammalian target of rapamycin |
| mTORC | mTOR complex |
| mg | milligram |
| MAPK | mitogen-activated protein kinase |
| MyD88 | Myeloid differentiation primary response 88 |
| ng | nanogram |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOD2 | nucleotide-oligonucleotide-domain 2 |
| pDCs | plasmacytoid dendritic cells |
| PI3K-γ | phosphatidylinositol 3-kinase-γ |
| SIGIRR | Single Ig IL-1-related receptor |
| SPF | Specific pathogen-free |
| T6SS | type VI secretion system |
| TIR | toll-Interleukin receptor domain |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TRAM | TRIF-related adapter molecule |
| TRIF | TIR-domain-containing adapter inducing interferon-β |
References
- Young, K.T.; Davis, L.M.; Dirita, V.J. Campylobacter jejuni: Molecular biology and pathogenesis. Nat. Rev. Microbiol. 2007, 5, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Nachamkin, I.; Szymanski, C.M.; Blaser, M.J. Campylobacter; ASM Press: Atlanta, GA, USA, 2008. [Google Scholar]
- Poly, F.; Guerry, P. Pathogenesis of Campylobacter. Curr. Opin. Gastroenterol. 2008, 24, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Krogfelt, K.A.; Cawthraw, S.A.; van Pelt, W.; Wagenaar, J.A.; Owen, R.J. Host-pathogen interactions in Campylobacter infections: The host perspective. Clin. Microbiol. Rev. 2008, 21, 505–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epps, S.V.; Harvey, R.B.; Hume, M.E.; Phillips, T.D.; Anderson, R.C.; Nisbet, D.J. Foodborne Campylobacter: Infections, metabolism, pathogenesis and reservoirs. Int. J. Environ. Res. Public Health 2013, 10, 6292–6304. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.C.; Jinneman, K.C.; Neal-McKinney, J.; Wu, W.H.; Rice, D.H. Simultaneous Identification of Campylobacter jejuni, Campylobacter coli, and Campylobacter lari with SmartCycler-Based Multiplex Quantitative Polymerase Chain Reaction. Foodborne Pathog. Dis. 2017, 14, 371–378. [Google Scholar] [CrossRef]
- Igwaran, A.; Okoh, A.I. Human campylobacteriosis: A public health concern of global importance. Heliyon 2019, 5, e02814. [Google Scholar] [CrossRef]
- EFSA. The Community Summary Report on antimicrobial resistance in zoonotic and indicator bacteria from animals and food in the European Union in 2008. EFSA J. 2010, 8, 1658. [Google Scholar] [CrossRef] [Green Version]
- Backert, S.; Tegtmeyer, N.; Cróinín, T.Ó.; Boehm, M.; Heimesaat, M.M. Chapter 1—Human campylobacteriosis. In Campylobacter; Klein, G., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 1–25. [Google Scholar] [CrossRef]
- Davis, L.; DiRita, V. Growth and laboratory maintenance of Campylobacter jejuni. Curr. Protoc. Microbiol. 2008, 8, 8A.1.1–8A.1.7. [Google Scholar] [CrossRef] [Green Version]
- Bolton, D.J. Campylobacter virulence and survival factors. Food Microbiol. 2015, 48, 99–108. [Google Scholar] [CrossRef]
- de Zoete, M.R.; Keestra, A.M.; Roszczenko, P.; van Putten, J.P. Activation of human and chicken toll-like receptors by Campylobacter spp. Infect. Immun. 2010, 78, 1229–1238. [Google Scholar] [CrossRef] [Green Version]
- Ellström, P.; Hansson, I.; Nilsson, A.; Rautelin, H.; Engvall, E.O. Lipooligosaccharide locus classes and putative virulence genes among chicken and human Campylobacter jejuni isolates. BMC Microbiol. 2016, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Hermans, D.; Pasmans, F.; Messens, W.; Martel, A.; Van Immerseel, F.; Rasschaert, G.; Heyndrickx, M.; Van Deun, K.; Haesebrouck, F. Poultry as a host for the zoonotic pathogen Campylobacter jejuni. Vector Borne Zoonotic Dis. 2012, 12, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.A.; Mehra, R.K.; Carrington, S.D.; Hickey, R.M. The food glycome: A source of protection against pathogen colonization in the gastrointestinal tract. Int. J. Food Microbiol. 2010, 142, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Havelaar, A.H.; van Pelt, W.; Ang, C.W.; Wagenaar, J.A.; van Putten, J.P.; Gross, U.; Newell, D.G. Immunity to Campylobacter: Its role in risk assessment and epidemiology. Crit. Rev. Microbiol. 2009, 35, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ó Cróinín, T.; Backert, S. Host epithelial cell invasion by Campylobacter jejuni: Trigger or zipper mechanism? Front. Cell. Infect. Microbiol. 2012, 2, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kist, M.; Bereswill, S. Campylobacter jejuni. Contrib. Microbiol. 2001, 8, 150–165. [Google Scholar] [PubMed]
- Mousavi, S.; Schmidt, A.-M.; Escher, U.; Kittler, S.; Kehrenberg, C.; Thunhorst, E.; Bereswill, S.; Heimesaat, M.M. Carvacrol ameliorates acute campylobacteriosis in a clinical murine infection model. Gut Pathog. 2020, 12, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, S.; Escher, U.; Thunhorst, E.; Kittler, S.; Kehrenberg, C.; Bereswill, S.; Heimesaat, M.M. Vitamin C alleviates acute enterocolitis in Campylobacter jejuni infected mice. Sci. Rep. 2020, 10, 2921. [Google Scholar] [CrossRef] [Green Version]
- Allos, B.M. Association between Campylobacter infection and Guillain-Barre syndrome. J. Infect. Dis. 1997, 176, S125–S128. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, N.P.; Kuijf, M.L.; Ang, C.W.; Schiellerup, P.; Krogfelt, K.A.; Jacobs, B.C.; van Belkum, A.; Endtz, H.P.; Bergman, M.P. Sialylation of Campylobacter jejuni lipo-oligosaccharides is associated with severe gastro-enteritis and reactive arthritis. Microbes Infect. 2009, 11, 988–994. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Castano-Rodriguez, N.; Mitchell, H.M.; Man, S.M. Global Epidemiology of Campylobacter Infection. Clin. Microbiol. Rev. 2015, 28, 687–720. [Google Scholar] [CrossRef] [Green Version]
- Naito, M.; Frirdich, E.; Fields, J.A.; Pryjma, M.; Li, J.; Cameron, A.; Gilbert, M.; Thompson, S.A.; Gaynor, E.C. Effects of sequential Campylobacter jejuni 81–176 lipooligosaccharide core truncations on biofilm formation, stress survival, and pathogenesis. J. Bacteriol. 2010, 192, 2182–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlyshev, A.V.; Ketley, J.M.; Wren, B.W. The Campylobacter jejuni glycome. FEMS Microbiol. Rev. 2005, 29, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Facciolà, A.; Riso, R.; Avventuroso, E.; Visalli, G.; Delia, S.A.; Laganà, P. Campylobacter: From microbiology to prevention. J. Prev. Med. Hyg. 2017, 58, E79–E92. [Google Scholar] [PubMed]
- Schmidt, A.M.; Escher, U.; Mousavi, S.; Boehm, M.; Backert, S.; Bereswill, S.; Heimesaat, M.M. Protease Activity of Campylobacter jejuni HtrA Modulates Distinct Intestinal and Systemic Immune Responses in Infected Secondary Abiotic IL-10 Deficient Mice. Front. Cell. Infect. Microbiol. 2019, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.-M.; Escher, U.; Mousavi, S.; Tegtmeyer, N.; Boehm, M.; Backert, S.; Bereswill, S.; Heimesaat, M.M. Immunopathological properties of the Campylobacter jejuni flagellins and the adhesin CadF as assessed in a clinical murine infection model. Gut Pathog. 2019, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Ketley, J.M.; Konkel, M.E. Campylobacter: Molecular and Cellular Biology; Horizon Scientific Press: Norfolk, UK, 2005. [Google Scholar]
- Lertsethtakarn, P.; Ottemann, K.M.; Hendrixson, D.R. Motility and chemotaxis in Campylobacter and Helicobacter. Ann. Rev. Microbiol. 2011, 65, 389–410. [Google Scholar] [CrossRef] [PubMed]
- Bleumink-Pluym, N.M.; van Alphen, L.B.; Bouwman, L.I.; Wosten, M.M.; van Putten, J.P. Identification of a functional type VI secretion system in Campylobacter jejuni conferring capsule polysaccharide sensitive cytotoxicity. PLoS Pathog. 2013, 9, e1003393. [Google Scholar] [CrossRef]
- Harrison, J.W.; Dung, T.T.; Siddiqui, F.; Korbrisate, S.; Bukhari, H.; Tra, M.P.; Hoang, N.V.; Carrique-Mas, J.; Bryant, J.; Campbell, J.I.; et al. Identification of possible virulence marker from Campylobacter jejuni isolates. Emerg. Infect. Dis. 2014, 20, 1026–1029. [Google Scholar] [CrossRef] [Green Version]
- Agnetti, J.; Seth-Smith, H.M.B.; Ursich, S.; Reist, J.; Basler, M.; Nickel, C.; Bassetti, S.; Ritz, N.; Tschudin-Sutter, S.; Egli, A. Clinical impact of the type VI secretion system on virulence of Campylobacter species during infection. BMC Infect. Dis. 2019, 19, 237. [Google Scholar] [CrossRef]
- Backert, S.; Boehm, M.; Wessler, S.; Tegtmeyer, N. Transmigration route of Campylobacter jejuni across polarized intestinal epithelial cells: Paracellular, transcellular or both? Cell Commun. Signal. CCS 2013, 11, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Spreeuwel, J.P.; Duursma, G.C.; Meijer, C.J.; Bax, R.; Rosekrans, P.C.; Lindeman, J. Campylobacter colitis: Histological immunohistochemical and ultrastructural findings. Gut 1985, 26, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bras, A.M.; Ketley, J.M. Transcellular translocation of Campylobacter jejuni across human polarised epithelial monolayers. FEMS Microbiol. Lett. 1999, 179, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Bucker, R.; Krug, S.M.; Moos, V.; Bojarski, C.; Schweiger, M.R.; Kerick, M.; Fromm, A.; Janssen, S.; Fromm, M.; Hering, N.A.; et al. Campylobacter jejuni impairs sodium transport and epithelial barrier function via cytokine release in human colon. Mucosal Immunol. 2018, 11, 575–577. [Google Scholar] [CrossRef] [Green Version]
- Mandrell, R.E.; Griffiss, J.M.; Macher, B.A. Lipooligosaccharides (LOS) of Neisseria gonorrhoeae and Neisseria meningitidis have components that are immunochemically similar to precursors of human blood group antigens. Carbohydrate sequence specificity of the mouse monoclonal antibodies that recognize crossreacting antigens on LOS and human erythrocytes. J. Exp. Med. 1988, 168, 107–126. [Google Scholar] [CrossRef]
- Louwen, R.; Heikema, A.; van Belkum, A.; Ott, A.; Gilbert, M.; Ang, W.; Endtz, H.P.; Bergman, M.P.; Nieuwenhuis, E.E. The sialylated lipooligosaccharide outer core in Campylobacter jejuni is an important determinant for epithelial cell invasion. Infect. Immun. 2008, 76, 4431–4438. [Google Scholar] [CrossRef] [Green Version]
- Perera, V.N.; Nachamkin, I.; Ung, H.; Patterson, J.H.; McConville, M.J.; Coloe, P.J.; Fry, B.N. Molecular mimicry in Campylobacter jejuni: Role of the lipo-oligosaccharide core oligosaccharide in inducing anti-ganglioside antibodies. FEMS Immunol. Med. Microbiol. 2007, 50, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Meyer, B.; Hanel, I.; Hotzel, H. Comparison of lipooligosaccharide biosynthesis genes of Campylobacter jejuni strains with varying abilities to colonize the chicken gut and to invade Caco-2 cells. J. Med. Microbiol. 2007, 56, 1589–1594. [Google Scholar] [CrossRef] [Green Version]
- Zarantonelli, M.L.; Huerre, M.; Taha, M.-K.; Alonso, J.-M. Differential role of lipooligosaccharide of Neisseria meningitidis in virulence and inflammatory response during respiratory infection in mice. Infect. Immun. 2006, 74, 5506–5512. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Bereswill, S. Murine infection models for the investigation of Campylobacter jejuni--host interactions and pathogenicity. Berl. Munch. Tierarztl. Wochenschr. 2015, 128, 98–103. [Google Scholar]
- Warren, H.S.; Fitting, C.; Hoff, E.; Adib-Conquy, M.; Beasley-Topliffe, L.; Tesini, B.; Liang, X.; Valentine, C.; Hellman, J.; Hayden, D.; et al. Resilience to bacterial infection: Difference between species could be due to proteins in serum. J. Infect. Dis. 2010, 201, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrell, N.; Wren, B.W. The second century of Campylobacter research: Recent advances, new opportunities and old problems. Curr. Opin. Infect. Dis. 2007, 20, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Bereswill, S.; Fischer, A.; Plickert, R.; Haag, L.M.; Otto, B.; Kuhl, A.A.; Dasti, J.I.; Zautner, A.E.; Munoz, M.; Loddenkemper, C.; et al. Novel murine infection models provide deep insights into the “menage a trois” of Campylobacter jejuni, microbiota and host innate immunity. PLoS ONE 2011, 6, e20953. [Google Scholar] [CrossRef]
- Fiebiger, U.; Bereswill, S.; Heimesaat, M.M. Dissecting the Interplay between Intestinal Microbiota and Host Immunity in Health and Disease: Lessons Learned from Germfree and Gnotobiotic Animal Models. Eur. J. Microbiol. Immunol. 2016, 6, 253–271. [Google Scholar] [CrossRef] [Green Version]
- Masanta, W.O.; Heimesaat, M.M.; Bereswill, S.; Tareen, A.M.; Lugert, R.; Groß, U.; Zautner, A.E. Modification of intestinal microbiota and its consequences for innate immune response in the pathogenesis of campylobacteriosis. Clin. Dev. Immunol. 2013, 2013, 526860. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Mrazek, K.; Bereswill, S. Murine Fecal Microbiota Transplantation Alleviates Intestinal and Systemic Immune Responses in Campylobacter jejuni Infected Mice Harboring a Human Gut Microbiota. Front. Immunol. 2019, 10, 2272. [Google Scholar] [CrossRef]
- Haag, L.M.; Fischer, A.; Otto, B.; Plickert, R.; Kuhl, A.A.; Gobel, U.B.; Bereswill, S.; Heimesaat, M.M. Campylobacter jejuni induces acute enterocolitis in gnotobiotic IL-10−/− mice via Toll-like-receptor-2 and -4 signaling. PLoS ONE 2012, 7, e40761. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Grundmann, U.; Alutis, M.E.; Fischer, A.; Bereswill, S. Absence of Nucleotide-Oligomerization-Domain-2 Is Associated with Less Distinct Disease in Campylobacter jejuni Infected Secondary Abiotic IL-10 Deficient Mice. Front. Cell. Infect. Microbiol. 2017, 7, 322. [Google Scholar] [CrossRef]
- Heimesaat, M.M.; Lugert, R.; Fischer, A.; Alutis, M.; Kuhl, A.A.; Zautner, A.E.; Tareen, A.M.; Gobel, U.B.; Bereswill, S. Impact of Campylobacter jejuni cj0268c knockout mutation on intestinal colonization, translocation, and induction of immunopathology in gnotobiotic IL-10 deficient mice. PLoS ONE 2014, 9, e90148. [Google Scholar] [CrossRef]
- Giallourou, N.; Medlock, G.L.; Bolick, D.T.; Medeiros, P.H.; Ledwaba, S.E.; Kolling, G.L.; Tung, K.; Guerry, P.; Swann, J.R.; Guerrant, R.L. A novel mouse model of Campylobacter jejuni enteropathy and diarrhea. PLoS Pathog. 2018, 14, e1007083. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; Ries, J.; Vermeulen, J.; Yang, H.; Sham, H.P.; Crowley, S.M.; Badayeva, Y.; Turvey, S.E.; Gaynor, E.C.; Li, X.; et al. A novel mouse model of Campylobacter jejuni gastroenteritis reveals key pro-inflammatory and tissue protective roles for Toll-like receptor signaling during infection. PLoS Pathog. 2014, 10, e1004264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellström, P.; Feodoroff, B.; Hänninen, M.L.; Rautelin, H. Lipooligosaccharide locus class of Campylobacter jejuni: Sialylation is not needed for invasive infection. Clin. Microbiol. Infect. 2014, 20, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfield, L.S.; Bell, J.A.; Wilson, D.L.; Murphy, A.J.; Elsheikha, H.M.; Rathinam, V.A.; Fierro, B.R.; Linz, J.E.; Young, V.B. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect. Immun. 2007, 75, 1099–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazmi, S.U.; Roberson, B.S.; Stern, N.J. Animal-passed, virulence-enhanced Campylobacter jejuni causes enteritis in neonatal mice. Curr. Microbiol. 1984, 11, 159–164. [Google Scholar] [CrossRef]
- Haag, L.M.; Fischer, A.; Otto, B.; Grundmann, U.; Kuhl, A.A.; Gobel, U.B.; Bereswill, S.; Heimesaat, M.M. Campylobacter jejuni infection of infant mice: Acute enterocolitis is followed by asymptomatic intestinal and extra-intestinal immune responses. Eur. J. Microbiol. Immunol. 2012, 2, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Yrios, J.W.; Balish, E. Colonization and infection of athymic and euthymic germfree mice by Campylobacter jejuni and Campylobacter fetus subsp. fetus. Infect. Immun. 1986, 53, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Yrios, J.W.; Balish, E. Pathogenesis of Campylobacter spp. in athymic and euthymic germfree mice. Infect. Immun. 1986, 53, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Otto, B.; Haag, L.M.; Fischer, A.; Plickert, R.; Kuhl, A.A.; Gobel, U.B.; Heimesaat, M.M.; Bereswill, S. Campylobacter jejuni induces extra-intestinal immune responses via Toll-like-receptor-4 signaling in conventional IL-10 deficient mice with chronic colitis. Eur. J. Microbiol. Immunol. 2012, 2, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Liu, B.; Sartor, R.B.; Jobin, C. Phosphatidylinositol 3-kinase-gamma signaling promotes Campylobacter jejuni-induced colitis through neutrophil recruitment in mice. J. Immunol. 2013, 190, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Lorne, E.; Zhao, X.; Zmijewski, J.W.; Liu, G.; Park, Y.J.; Tsuruta, Y.; Abraham, E. Participation of mammalian target of rapamycin complex 1 in Toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am. J. Respir. Cell. Mol. Biol. 2009, 41, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Alutis, M.; Grundmann, U.; Fischer, A.; Tegtmeyer, N.; Bohm, M.; Kuhl, A.A.; Gobel, U.B.; Backert, S.; Bereswill, S. The role of serine protease HtrA in acute ulcerative enterocolitis and extra-intestinal immune responses during Campylobacter jejuni infection of gnotobiotic IL-10 deficient mice. Front. Cell. Infect. Microbiol. 2014, 4, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, M.J.; Berkowitz, I.D.; LaForce, F.M.; Cravens, J.; Reller, L.B.; Wang, W.L. Campylobacter enteritis: Clinical and epidemiologic features. Ann. Intern. Med. 1979, 91, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Gradel, K.O.; Nielsen, H.L.; Schonheyder, H.C.; Ejlertsen, T.; Kristensen, B.; Nielsen, H. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology 2009, 137, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, S.; Kapadnis, B. Virulence factors of Campylobacter. Internet J. Microbiol. 2007, 3, 1–7. [Google Scholar]
- Ketley, J.M. Pathogenesis of enteric infection by Campylobacter. Microbiology (Read. Engl.) 1997, 143, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.I.; Caldwell, M.B.; Lee, E.C.; Guerry, P.; Trust, T.J.; Ruiz-Palacios, G.M. Pathophysiology of Campylobacter enteritis. Microbiol. Rev. 1986, 50, 81–94. [Google Scholar] [CrossRef]
- Guerry, P.; Ewing, C.P.; Hickey, T.E.; Prendergast, M.M.; Moran, A.P. Sialylation of lipooligosaccharide cores affects immunogenicity and serum resistance of Campylobacter jejuni. Infect. Immun. 2000, 68, 6656–6662. [Google Scholar] [CrossRef] [Green Version]
- Habib, I.; Louwen, R.; Uyttendaele, M.; Houf, K.; Vandenberg, O.; Nieuwenhuis, E.E.; Miller, W.G.; van Belkum, A.; De Zutter, L. Correlation between genotypic diversity, lipooligosaccharide gene locus class variation, and caco-2 cell invasion potential of Campylobacter jejuni isolates from chicken meat and humans: Contribution to virulotyping. Appl. Environ. Microbiol. 2009, 75, 4277–4288. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.; Godschalk, P.C.; Karwaski, M.F.; Ang, C.W.; van Belkum, A.; Li, J.; Wakarchuk, W.W.; Endtz, H.P. Evidence for acquisition of the lipooligosaccharide biosynthesis locus in Campylobacter jejuni GB11, a strain isolated from a patient with Guillain-Barre syndrome, by horizontal exchange. Infect. Immun. 2004, 72, 1162–1165. [Google Scholar] [CrossRef] [Green Version]
- Godschalk, P.C.; Kuijf, M.L.; Li, J.; St Michael, F.; Ang, C.W.; Jacobs, B.C.; Karwaski, M.F.; Brochu, D.; Moterassed, A.; Endtz, H.P.; et al. Structural characterization of Campylobacter jejuni lipooligosaccharide outer cores associated with Guillain-Barre and Miller Fisher syndromes. Infect. Immun. 2007, 75, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Riddle, M.S.; Guerry, P. Status of vaccine research and development for Campylobacter jejuni. Vaccine 2016, 34, 2903–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halpin, A.L.; Gu, W.; Wise, M.E.; Sejvar, J.J.; Hoekstra, R.M.; Mahon, B.E. Post-Campylobacter Guillain Barré Syndrome in the USA: Secondary analysis of surveillance data collected during the 2009–2010 novel Influenza A (H1N1) vaccination campaign. Epidemiol. Infect. 2018, 146, 1740–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.; Brisson, J.R.; Karwaski, M.F.; Michniewicz, J.; Cunningham, A.M.; Wu, Y.; Young, N.M.; Wakarchuk, W.W. Biosynthesis of ganglioside mimics in Campylobacter jejuni OH4384. Identification of the glycosyltransferase genes, enzymatic synthesis of model compounds, and characterization of nanomole amounts by 600-mhz (1)h and (13)c NMR analysis. J. Biol. Chem. 2000, 275, 3896–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, M.; Gilbert, M.; Takahashi, M.; Li, J.; Koike, S.; Hirata, K.; Yuki, N. Comprehensive analysis of bacterial risk factors for the development of Guillain-Barre syndrome after Campylobacter jejuni enteritis. J. Infect. Dis. 2006, 193, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Louwen, R.; Nieuwenhuis, E.E.; van Marrewijk, L.; Horst-Kreft, D.; de Ruiter, L.; Heikema, A.P.; van Wamel, W.J.; Wagenaar, J.A.; Endtz, H.P.; Samsom, J.; et al. Campylobacter jejuni translocation across intestinal epithelial cells is facilitated by ganglioside-like lipooligosaccharide structures. Infect. Immun. 2012, 80, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Golec, M. Cathelicidin LL-37: LPS-neutralizing, pleiotropic peptide. Ann. Agric. Environ. Med. 2007, 14, 1–4. [Google Scholar]
- Houliston, R.S.; Vinogradov, E.; Dzieciatkowska, M.; Li, J.; St Michael, F.; Karwaski, M.-F.; Brochu, D.; Jarrell, H.C.; Parker, C.T.; Yuki, N.; et al. Lipooligosaccharide of Campylobacter jejuni: Similarity with multiple types of mammalian glycans beyond gangliosides. J. Biol. Chem. 2011, 286, 12361–12370. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.; Karwaski, M.F.; Bernatchez, S.; Young, N.M.; Taboada, E.; Michniewicz, J.; Cunningham, A.M.; Wakarchuk, W.W. The genetic bases for the variation in the lipo-oligosaccharide of the mucosal pathogen, Campylobacter jejuni. Biosynthesis of sialylated ganglioside mimics in the core oligosaccharide. J. Biol. Chem. 2002, 277, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Guerry, P.; Szymanski, C.M.; Prendergast, M.M.; Hickey, T.E.; Ewing, C.P.; Pattarini, D.L.; Moran, A.P. Phase variation of Campylobacter jejuni 81–176 lipooligosaccharide affects ganglioside mimicry and invasiveness in vitro. Infect. Immun. 2002, 70, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol Rev 1992, 56, 395–411. [Google Scholar] [CrossRef]
- Oh, E.; Jeon, B. Contribution of surface polysaccharides to the resistance of Campylobacter jejuni to antimicrobial phenolic compounds. J. Antibiot. 2015, 68, 591. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, N.J.; Moran, A.P.; Millar, L.A.; Prendergast, M.M.; Ketley, J.M. Characterization of the Campylobacter jejuni heptosyltransferase II gene, waaF, provides genetic evidence that extracellular polysaccharide is lipid A core independent. J. Bacteriol. 2002, 184, 2100–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungi, T.W.; Farhat, K.; Burgener, I.A.; Werling, D. Toll-like receptors in domestic animals. Cell Tissue Res. 2011, 343, 107–120. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Fitzgerald, K.A.; Bowie, A.G. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003, 24, 286–290. [Google Scholar] [CrossRef]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.K.; Appledorn, D.M.; Hoag, K.A.; Amalfitano, A.; Mansfield, L.S. Campylobacter jejuni-induced activation of dendritic cells involves cooperative signaling through Toll-like receptor 4 (TLR4)-MyD88 and TLR4-TRIF axes. Infect. Immun. 2009, 77, 2499–2507. [Google Scholar] [CrossRef] [Green Version]
- van Mourik, A.; Steeghs, L.; van Laar, J.; Meiring, H.D.; Hamstra, H.J.; van Putten, J.P.; Wosten, M.M. Altered linkage of hydroxyacyl chains in lipid A of Campylobacter jejuni reduces TLR4 activation and antimicrobial resistance. J. Biol. Chem. 2010, 285, 15828–15836. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, H.N.; John, C.M.; Naz, N.; Gundogdu, O.; Dorrell, N.; Wren, B.W.; Jarvis, G.A.; Bajaj-Elliott, M. Campylobacter jejuni lipooligosaccharide sialylation, phosphorylation, and amide/ester linkage modifications fine-tune human Toll-like receptor 4 activation. J. Biol. Chem. 2013, 288, 19661–19672. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M. Role of Toll-like receptors in Helicobacter pylori infection and immunity. World J. Gastrointest. Pathophysiol. 2014, 5, 133–146. [Google Scholar] [CrossRef]
- Kuijf, M.L.; Samsom, J.N.; van Rijs, W.; Bax, M.; Huizinga, R.; Heikema, A.P.; van Doorn, P.A.; van Belkum, A.; van Kooyk, Y.; Burgers, P.C.; et al. TLR4-mediated sensing of Campylobacter jejuni by dendritic cells is determined by sialylation. J. Immunol. 2010, 185, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Ketloy, C.; Engering, A.; Srichairatanakul, U.; Limsalakpetch, A.; Yongvanitchit, K.; Pichyangkul, S.; Ruxrungtham, K. Expression and function of Toll-like receptors on dendritic cells and other antigen presenting cells from non-human primates. Vet. Immunol. Immunopathol. 2008, 125, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, A.M.; Ernst, R.K.; Tsai, J.H.; Wilson, C.B.; Miller, S.I. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 2002, 3, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Bosisio, D.; Polentarutti, N.; D’Amico, G.; Stoppacciaro, A.; Mancinelli, R.; van’t Veer, C.; Penton-Rol, G.; Ruco, L.P.; Allavena, P.; et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: Selective expression of TLR3 in dendritic cells. J. Immunol. 2000, 164, 5998–6004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, F.; Akashi, S.; Sakao, Y.; Sato, S.; Kawai, T.; Matsumoto, M.; Nakanishi, K.; Kimoto, M.; Miyake, K.; Takeda, K.; et al. Cutting edge: Endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 2000, 164, 3476–3479. [Google Scholar] [CrossRef] [Green Version]
- Molteni, M.; Bosi, A.; Rossetti, C. Natural Products with Toll-Like Receptor 4 Antagonist Activity. Int. J. Inflamm. 2018, 2018, 2859135. [Google Scholar] [CrossRef] [Green Version]
- Lobo de Sa, F.D.; Butkevych, E.; Nattramilarasu, P.K.; Fromm, A.; Mousavi, S.; Moos, V.; Golz, J.C.; Stingl, K.; Kittler, S.; Seinige, D.; et al. Curcumin Mitigates Immune-Induced Epithelial Barrier Dysfunction by Campylobacter jejuni. Int. J. Mol. Sci. 2019, 20, 4830. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ghosh, S.S.; Ghosh, S. Curcumin improves intestinal barrier function: Modulation of intracellular signaling, and organization of tight junctions. Am. J. Physiol. Cell Physiol. 2017, 312, C438–C445. [Google Scholar] [CrossRef]
- Youn, H.S.; Saitoh, S.I.; Miyake, K.; Hwang, D.H. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem. Pharmacol. 2006, 72, 62–69. [Google Scholar] [CrossRef]
- Senior, N.J.; Bagnall, M.C.; Champion, O.L.; Reynolds, S.E.; La Ragione, R.M.; Woodward, M.J.; Salguero, F.J.; Titball, R.W. Galleria mellonella as an infection model for Campylobacter jejuni virulence. J. Med. Microbiol. 2011, 60, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Babakhani, F.K.; Bradley, G.A.; Joens, L.A. Newborn piglet model for campylobacteriosis. Infect. Immun. 1993, 61, 3466–3475. [Google Scholar] [CrossRef] [Green Version]
- Vitovec, J.; Koudela, B.; Sterba, J.; Tomancova, I.; Matyas, Z.; Vladik, P. The gnotobiotic piglet as a model for the pathogenesis of Campylobacter jejuni infection. Zent. Bakteriol. Int. J. Med. Microbiol. 1989, 271, 91–103. [Google Scholar] [CrossRef]
- Mansfield, L.S.; Gauthier, D.T.; Abner, S.R.; Jones, K.M.; Wilder, S.R.; Urban, J.F. Enhancement of disease and pathology by synergy of Trichuris suis and Campylobacter jejuni in the colon of immunologically naive swine. Am. J. Trop. Med. Hyg. 2003, 68, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G.; Ackerman, J.I.; Taylor, N.; Claps, M.; Murphy, J.C. Campylobacter jejuni infection in the ferret: An animal model of human campylobacteriosis. Am. J. Vet. Res. 1987, 48, 85–90. [Google Scholar] [PubMed]
- Bereswill, S.; Plickert, R.; Fischer, A.; Kühl, A.A.; Loddenkemper, C.; Batra, A.; Siegmund, B.; Göbel, U.B.; Heimesaat, M.M. What you eat is what you get: Novel Campylobacter models in the quadrangle relationship between nutrition, obesity, microbiota and susceptibility to infection. Eur. J. Microbiol. Immunol. 2011, 1, 237–248. [Google Scholar] [CrossRef]
- Haag, L.M.; Fischer, A.; Otto, B.; Plickert, R.; Kuhl, A.A.; Gobel, U.B.; Bereswill, S.; Heimesaat, M.M. Intestinal microbiota shifts towards elevated commensal Escherichia coli loads abrogate colonization resistance against Campylobacter jejuni in mice. PLoS ONE 2012, 7, e35988. [Google Scholar] [CrossRef]
- Lotz, M.; Gütle, D.; Walther, S.; Ménard, S.; Bogdan, C.; Hornef, M.W. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J. Exp. Med. 2006, 203, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.L.; Gold, M.J.; Hartmann, M.; Willing, B.P.; Thorson, L.; Wlodarska, M.; Gill, N.; Blanchet, M.R.; Mohn, W.W.; McNagny, K.M.; et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012, 13, 440–447. [Google Scholar] [CrossRef]
- Stahl, M.; Vallance, B.A. Insights into Campylobacter jejuni colonization of the mammalian intestinal tract using a novel mouse model of infection. Gut Microbes 2015, 6, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Gulen, M.F.; Qin, J.; Yao, J.; Bulek, K.; Kish, D.; Altuntas, C.Z.; Wald, D.; Ma, C.; Zhou, H.; et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 2007, 26, 461–475. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.A.; Care, A.S.; Skinner, R.J. Interleukin 10 regulates inflammatory cytokine synthesis to protect against lipopolysaccharide-induced abortion and fetal growth restriction in mice. Biol. Reprod. 2007, 76, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.A.; Skinner, R.J.; Care, A.S. Essential role for IL-10 in resistance to lipopolysaccharide-induced preterm labor in mice. J. Immunol. 2006, 177, 4888–4896. [Google Scholar] [CrossRef] [PubMed]
- Emoto, M.; Emoto, Y.; Brinkmann, V.; Miyamoto, M.; Yoshizawa, I.; Staber, M.; van Rooijen, N.; Hamann, A.; Kaufmann, S.H. Increased resistance of LFA-1-deficient mice to lipopolysaccharide-induced shock/liver injury in the presence of TNF-alpha and IL-12 is mediated by IL-10: A novel role for LFA-1 in the regulation of the proinflammatory and anti-inflammatory cytokine balance. J. Immunol. 2003, 171, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Lippert, E.; Karrasch, T.; Sun, X.; Allard, B.; Herfarth, H.H.; Threadgill, D.; Jobin, C. Gnotobiotic IL-10; NF-kappaB mice develop rapid and severe colitis following Campylobacter jejuni infection. PLoS ONE 2009, 4, e7413. [Google Scholar] [CrossRef]
- Vikbladh, I. Studies on zinc in blood II. Scand. J. Clin. Lab. Invest. 1951, 3, 1–74. [Google Scholar]
- Auerbach, S. Zinc Content of Plasma, Blood, and Erythrocytes in Normal Subjects and in Patients with Hodgkin’s Disease and Various Hematologic Disorders. J. Lab. Clin. Med. 1965, 65, 628–637. [Google Scholar]
- Pekarek, R.S.; Beisel, W.R. Effect of endotoxin on serum zinc concentrations in the rat. Appl. Microbiol. 1969, 18, 482–484. [Google Scholar] [CrossRef] [Green Version]
- Snyder, S.L.; Walker, R.I. Inhibition of lethality in endotoxin-challenged mice treated with zinc chloride. Infect. Immun. 1976, 13, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Thambiayya, K.; Wasserloos, K.; Kagan, V.E.; Stoyanovsky, D.; Pitt, B.R. A critical role for increased labile zinc in reducing sensitivity of cultured sheep pulmonary artery endothelial cells to LPS-induced apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1287–L1295. [Google Scholar] [CrossRef] [Green Version]
- Rink, L.; Haase, H. Zinc homeostasis and immunity. Trends Immunol. 2007, 28, 1–4. [Google Scholar] [CrossRef]
- Haase, H.; Rink, L. Functional significance of zinc-related signaling pathways in immune cells. Ann. Rev. Nutr. 2009, 29, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.F.; Black, R.E. Zinc and the risk for infectious disease. Annu. Rev. Nutr. 2004, 24, 255–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Zhao, M.; Chen, X.; Zhang, Y.; Wang, H.; Huang, Y.Y.; Wang, Z.; Zhang, Z.H.; Zhang, C.; Xu, D.X. Zinc supplementation during pregnancy protects against lipopolysaccharide-induced fetal growth restriction and demise through its anti-inflammatory effect. J. Immunol. 2012, 189, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Ohata, S.; Moriyama, C.; Yamashita, A.; Nishida, T.; Kusumoto, C.; Mochida, S.; Minami, Y.; Nakada, J.; Shomori, K.; Inagaki, Y.; et al. Polaprezinc Protects Mice against Endotoxin Shock. J. Clin. Biochem. Nutr. 2010, 46, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Suwendi, E.; Iwaya, H.; Lee, J.-S.; Hara, H.; Ishizuka, S. Zinc deficiency induces dysregulation of cytokine productions in an experimental colitis of rats. Biomed. Res. 2012, 33, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Satsangi, J.; Wolstencroft, R.A.; Cason, J.; Ainley, C.C.; Dumonde, D.C.; Thompson, R.P. Interleukin 1 in Crohn’s disease. Clin. Exp. Immunol. 1987, 67, 594–605. [Google Scholar]
- Sarabi Asiabar, A.; Asadzadeh Aghdaei, H.; Zamani, S.; Bokaie, S.; Zali, M.R.; Feizabadi, M.M. Molecular detection of Campylobacter jejuni in patients with Crohn’s disease in Iran. Med. J. Islamic Repub. Iran 2019, 33, 76. [Google Scholar] [CrossRef]
- Bereswill, S.; Alter, T. Editorial. Berl. Munch. Tierarztl. Wochenschr. 2015, 128, 89. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.; Scheffold, A.; Rutz, S. Functions and regulation of T cell-derived interleukin-10. Semin. Immunol. 2019, 44, 101344. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.P.; Amiel, E. Regulation of Dendritic Cell Immune Function and Metabolism by Cellular Nutrient Sensor Mammalian Target of Rapamycin (mTOR). Front. Immunol. 2018, 9, 3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, M.; Hoshii, T.; Fujii, H.; Koyasu, S.; Hirao, A.; Matsuda, S. Cutting edge: Mtorc1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J. Immunol. 2012, 188, 4736–4740. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Threadgill, D.; Jobin, C. Campylobacter jejuni induces colitis through activation of mammalian target of rapamycin signaling. Gastroenterology 2012, 142, 86–95. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.á.; Lior, H. A new heat-labile cytolethal distending toxin (CLDT) produced by Campylobacter spp. Microb. Pathog. 1988, 4, 115–126. [Google Scholar] [CrossRef]
- Hickey, T.E.; McVeigh, A.L.; Scott, D.A.; Michielutti, R.E.; Bixby, A.; Carroll, S.A.; Bourgeois, A.L.; Guerry, P. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect. Immun. 2000, 68, 6535–6541. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Jobin, C. Nucleotide-binding oligomerization domain-containing protein 2 controls host response to Campylobacter jejuni in Il10−/− mice. J. Infect. Dis. 2014, 210, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Bereswill, S.; Grundmann, U.; Alutis, M.E.; Fischer, A.; Heimesaat, M.M. Campylobacter jejuni infection of conventionally colonized mice lacking nucleotide-oligomerization-domain-2. Gut Pathog. 2017, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Bereswill, S.; Grundmann, U.; Alutis, M.E.; Fischer, A.; Kuhl, A.A.; Heimesaat, M.M. Immune responses upon Campylobacter jejuni infection of secondary abiotic mice lacking nucleotide-oligomerization-domain-2. Gut Pathog. 2017, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Heimesaat, M.M.; Grundmann, U.; Alutis, M.E.; Fischer, A.; Bereswill, S. Microbiota Composition and Immune Responses During Campylobacter Jejuni Infection in Conventionally Colonized IL-10(−/−) Mice Lacking Nucleotide Oligomerization Domain 2. Eur. J. Microbiol. Immunol. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimesaat, M.M.; Grundmann, U.; Alutis, M.E.; Fischer, A.; Bereswill, S. Small Intestinal Pro-Inflammatory Immune Responses Following Campylobacter Jejuni Infection of Secondary Abiotic IL-10(−/−) Mice Lacking Nucleotide-Oligomerization-Domain-2. Eur. J. Microbiol. Immunol. 2017, 7, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.T.; Mansfield, L.S. Effects of antibiotic resistance (AR) and microbiota shifts on Campylobacter jejuni-mediated diseases. Anim. Health Res. Rev. 2017, 18, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.T.; Brakel, K.A.; Bell, J.A.; Bejcek, C.E.; Gilpin, T.; Brudvig, J.M.; Mansfield, L.S. Transplanted human fecal microbiota enhanced Guillain Barre syndrome autoantibody responses after Campylobacter jejuni infection in C57BL/6 mice. Microbiome 2017, 5, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.T.; Bell, J.A.; Bejcek, C.E.; Malik, A.; Mansfield, L.S. An antibiotic depleted microbiome drives severe Campylobacter jejuni-mediated Type 1/17 colitis, Type 2 autoimmunity and neurologic sequelae in a mouse model. J. Neuroimmunol. 2019, 337, 577048. [Google Scholar] [CrossRef] [PubMed]
- Layunta, E.; Latorre, E.; Forcen, R.; Grasa, L.; Castro, M.; Arias, M.A.; Alcalde, A.I.; Mesonero, J.E. NOD2 Modulates Serotonin Transporter and Interacts with TLR2 and TLR4 in Intestinal Epithelial Cells. Cell. Physiol. Biochem. 2018, 47, 1217–1229. [Google Scholar] [CrossRef]
- Martinez-Chamorro, A.; Moreno, A.; Gomez-Garcia, M.; Cabello, M.J.; Martin, J.; Lopez-Nevot, M.A. Epistatic interaction between TLR4 and NOD2 in patients with Crohn’s Disease: Relation with risk and phenotype in a Spanish cohort. Immunobiology 2016, 221, 927–933. [Google Scholar] [CrossRef]
- Fritz, J.H.; Girardin, S.E.; Fitting, C.; Werts, C.; Mengin-Lecreulx, D.; Caroff, M.; Cavaillon, J.M.; Philpott, D.J.; Adib-Conquy, M. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur. J. Immunol. 2005, 35, 2459–2470. [Google Scholar] [CrossRef]
- Tada, H.; Aiba, S.; Shibata, K.-I.; Ohteki, T.; Takada, H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect. Immun. 2005, 73, 7967–7976. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Ferwerda, G.; de Jong, D.J.; Jansen, T.; Jacobs, L.; Kramer, M.; Naber, T.H.J.; Drenth, J.P.H.; Girardin, S.E.; Jan Kullberg, B.; et al. Nucleotide-Binding Oligomerization Domain-2 Modulates Specific TLR Pathways for the Induction of Cytokine Release. J. Immunol. 2005, 174, 6518–6523. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zhao, Q.; Zheng, H.; Li, X.; Zhang, T.; Ma, X. A novel crosstalk between TLR4- and NOD2-mediated signaling in the regulation of intestinal inflammation. Sci. Rep. 2015, 5, 12018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullberg, B.J.; Ferwerda, G.; de Jong, D.J.; Drenth, J.P.; Joosten, L.A.; Van der Meer, J.W.; Netea, M.G. Crohn’s disease patients homozygous for the 3020insC NOD2 mutation have a defective NOD2/TLR4 cross-tolerance to intestinal stimuli. Immunology 2008, 123, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Asano, N.; Murray, P.J.; Ozato, K.; Tailor, P.; Fuss, I.J.; Kitani, A.; Strober, W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J. Clin. Investig. 2008, 118, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Kitani, A.; Murray, P.J.; Wakatsuki, Y.; Fuss, I.J.; Strober, W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity 2006, 25, 473–485. [Google Scholar] [CrossRef] [Green Version]
- Udden, S.M.N.; Peng, L.; Gan, J.L.; Shelton, J.M.; Malter, J.S.; Hooper, L.V.; Zaki, M.H. NOD2 Suppresses Colorectal Tumorigenesis via Downregulation of the TLR Pathways. Cell Rep. 2017, 19, 2756–2770. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.; Lobo de Sa, F.D.; Schulzke, J.D.; Bucker, R.; Bereswill, S.; Heimesaat, M.M. Vitamin D in Acute Campylobacteriosis-Results From an Intervention Study Applying a Clinical Campylobacter jejuni Induced Enterocolitis Model. Front. Immunol. 2019, 10, 2094. [Google Scholar] [CrossRef] [Green Version]
- Guerry, P. Campylobacter flagella: Not just for motility. Trends Microbiol. 2007, 15, 456–461. [Google Scholar] [CrossRef]
- Maue, A.C.; Poly, F.; Guerry, P. A capsule conjugate vaccine approach to prevent diarrheal disease caused by Campylobacter jejuni. Hum. Vaccines Immunother. 2014, 10, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Willis, L.M.; Whitfield, C. KpsC and KpsS are retaining 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) transferases involved in synthesis of bacterial capsules. Proc. Natl. Acad. Sci. USA 2013, 110, 20753–20758. [Google Scholar] [CrossRef] [Green Version]
- Jones, F.R.; Baqar, S.; Gozalo, A.; Nunez, G.; Espinoza, N.; Reyes, S.M.; Salazar, M.; Meza, R.; Porter, C.K.; Walz, S.E. New World monkey Aotus nancymae as a model for Campylobacter jejuni infection and immunity. Infect. Immun. 2006, 74, 790–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothaft, H.; Davis, B.; Lock, Y.Y.; Perez-Munoz, M.E.; Vinogradov, E.; Walter, J.; Coros, C.; Szymanski, C.M. Engineering the Campylobacter jejuni N-glycan to create an effective chicken vaccine. Sci. Rep. 2016, 6, 26511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dijk, A.; Herrebout, M.; Tersteeg-Zijderveld, M.H.; Tjeerdsma-van Bokhoven, J.L.; Bleumink-Pluym, N.; Jansman, A.J.; Veldhuizen, E.J.; Haagsman, H.P. Campylobacter jejuni is highly susceptible to killing by chicken host defense peptide cathelicidin-2 and suppresses intestinal cathelicidin-2 expression in young broilers. Vet. Microbiol. 2012, 160, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Achen, M.; Morishita, T.Y.; Ley, E.C. Shedding and colonization of Campylobacter jejuni in broilers from day-of-hatch to slaughter age. Avian Dis. 1998, 42, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Shreeve, J.E.; Toszeghy, M.; Pattison, M.; Newell, D.G. Sequential spread of Campylobacter infection in a multipen broiler house. Avian Dis. 2000, 44, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Meunier, M.; Guyard-Nicodeme, M.; Vigouroux, E.; Poezevara, T.; Beven, V.; Quesne, S.; Bigault, L.; Amelot, M.; Dory, D.; Chemaly, M. Promising new vaccine candidates against Campylobacter in broilers. PLoS ONE 2017, 12, e0188472. [Google Scholar] [CrossRef] [Green Version]
- Alemka, A.; Nothaft, H.; Zheng, J.; Szymanski, C.M. N-glycosylation of Campylobacter jejuni surface proteins promotes bacterial fitness. Infect. Immun. 2013, 81, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Nothaft, H.; Scott, N.E.; Vinogradov, E.; Liu, X.; Hu, R.; Beadle, B.; Fodor, C.; Miller, W.G.; Li, J.; Cordwell, S.J.; et al. Diversity in the protein N-glycosylation pathways within the Campylobacter genus. Mol. Cell. Proteom. MCP 2012, 11, 1203–1219. [Google Scholar] [CrossRef] [Green Version]
- Adler, H.E.; DaMassa, A.J. Toxicity of endotoxin to chicks. Avian Dis 1979, 23, 174–178. [Google Scholar] [CrossRef]
- Beery, J.T.; Hugdahl, M.B.; Doyle, M.P. Colonization of gastrointestinal tracts of chicks by Campylobacter jejuni. Appl. Environ. Microbiol. 1988, 54, 2365–2370. [Google Scholar] [CrossRef] [Green Version]
- Meinersmann, R.J.; Rigsby, W.E.; Stern, N.J.; Kelley, L.C.; Hill, J.E.; Doyle, M.P. Comparative study of colonizing and noncolonizing Campylobacter jejuni. Am. J. Vet. Res. 1991, 52, 1518–1522. [Google Scholar] [PubMed]
- Zhang, Q.; Sahin, O. Campylobacteriosis. Dis. Poult. 2020, 12, 754–769. [Google Scholar]
- Young, C.R.; Ziprin, R.L.; Hume, M.E.; Stanker, L.H. Dose response and organ invasion of day-of-hatch Leghorn chicks by different isolates of Campylobacter jejuni. Avian Dis. 1999, 118, 763–767. [Google Scholar] [CrossRef]
- Martich, G.; Danner, R.; Ceska, M.; Suffredini, A. Detection of interleukin 8 and tumor necrosis factor in normal humans after intravenous endotoxin: The effect of antiinflammatory agents. J. Exp. Med. 1991, 173, 1021–1024. [Google Scholar] [CrossRef] [Green Version]
- da Silva, A.M.T.; Kaulbach, H.C.; Chuidian, F.S.; Lambert, D.R.; Suffredini, A.F.; Danner, R.L. Shock and multiple-organ dysfunction after self-administration of Salmonella endotoxin. N. Engl. J. Med. 1993, 328, 1457–1460. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Strain | Intestinal Microbiota | Antibiotic Treatment/Specific Diet | Intestinal Colonization of C. jejuni | LOS Sensitized | Macroscopic Signs of Disease | Inflammatory Responses | Reference |
|---|---|---|---|---|---|---|---|
| C57BL/6 | SPF | No/No | Low | No | No | Elevated T-helper-1 cell responses in intestinal compartments | [56] |
| C57BL/6 | SPF | No/No | No | No | No | Intestinal epithelial cell apoptosis and proliferation/regeneration, moderate cellular and molecular inflammatory responses | [46] |
| C57BL/6 | No-SA | Yes/No | High | No | No | Intestinal epithelial cell apoptosis and proliferation/regeneration, pronounced cellular and molecular inflammatory responses | [46] |
| C57BL/6 | SA plus rHF | Yes/No | Moderate | No | No | Intestinal epithelial cell apoptosis and proliferation/regeneration, pronounced cellular and molecular inflammatory responses | [46] |
| BALB/c Neonatal | SPF | No/No | Moderate | Yes | Wasting, bloody diarrhea | ND | [57] |
| C57BL/6 Infant | SPF | No/No | Moderate | Yes | Wasting, bloody diarrhea (self-limiting) | Intestinal epithelial cell apoptosis and proliferation/regeneration, elevated pro-inflammatory mediators, extra-intestinal T cell infiltrates | [58] |
| C57BL/6 Zinc deficient | Depleted | Yes/Zinc depleted | High | Yes | Wasting, bloody diarrhea | Intestinal edema, pronounced neutrophilic infiltration, crypt hyperplasia, intestinal and systemic inflammatory mediator responses | [53] |
| Strain/Species | Genetic Modification | Intestinal Microflora | Antibiotic Treatment | LOS Sensitized | Macroscopic Signs of Disease | Inflammatory Responses | Reference |
|---|---|---|---|---|---|---|---|
| BALB/c | Athymic (nu/nu) Euthymic (+/nu) | No, GF | No | Yes | Transient diarrhea | Cecal shrinkage, accumulation of eosinophils in the lower intestinal mucosa and lamina propria, translocation of viable C. jejuni to MLN, spleen, liver and kidneys | [59,60] |
| C57BL/6 | IL-10−/− | SPF | No | Yes | Wasting, bloody diarrhea | Severe typhlocolitis and gross pathological changes in the gastrointestinal tract, increased immune cell responses and production of pro-inflammatory mediators | [56] |
| 129/SvEv | IL-10−/− | No, GF | No | Yes | Wasting, bloody diarrhea | Intestinal crypt abscesses and epithelial ulcerations. Increased mononuclear cells, neutrophils, and elevated pro-inflammatory mediators | [61] |
| 129/SvEv C57BL/6 | IL-10−/− | No, GF | No | Yes | Wasting, bloody diarrhea | Pronounced neutrophilic infiltration of the intestines; increased NF-κB activity and Il1β, Cxcl2, Il17a expression | [62,63] |
| C57BL/10 | IL-10−/− | No, SA | Yes | Yes | Wasting, bloody diarrhea | Increases in apoptotic intestinal epithelial cells. Pronounced innate and adaptive immune cell responses, elevated intestinal pro-inflammatory mediators. Translocation of viable C. jejuni to MLN, extra-intestinal including systemic inflammation | [50,64] |
| C57BL/6 | SIGIRR−/− | Modified | Yes | Yes | ND | Increases in intestinal granulocytes, goblet cell depletion, edema, elevated pro-inflammatory mediators, translocation of C. jejuni to subepithelial tissues | [54] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mousavi, S.; Bereswill, S.; Heimesaat, M.M. Novel Clinical Campylobacter jejuni Infection Models Based on Sensitization of Mice to Lipooligosaccharide, a Major Bacterial Factor Triggering Innate Immune Responses in Human Campylobacteriosis. Microorganisms 2020, 8, 482. https://doi.org/10.3390/microorganisms8040482
Mousavi S, Bereswill S, Heimesaat MM. Novel Clinical Campylobacter jejuni Infection Models Based on Sensitization of Mice to Lipooligosaccharide, a Major Bacterial Factor Triggering Innate Immune Responses in Human Campylobacteriosis. Microorganisms. 2020; 8(4):482. https://doi.org/10.3390/microorganisms8040482
Chicago/Turabian StyleMousavi, Soraya, Stefan Bereswill, and Markus M. Heimesaat. 2020. "Novel Clinical Campylobacter jejuni Infection Models Based on Sensitization of Mice to Lipooligosaccharide, a Major Bacterial Factor Triggering Innate Immune Responses in Human Campylobacteriosis" Microorganisms 8, no. 4: 482. https://doi.org/10.3390/microorganisms8040482