Functional Analysis of a Polluted River Microbiome Reveals a Metabolic Potential for Bioremediation

 ,
,
Abstract
:
1. Introduction
2. Material and Methods
2.1. Study Site, Sample Collection, and Chemical Parameters Measured
2.2. DNA Extraction and Sequencing
2.3. Bioinformatics Analysis
2.4. Biodegradative and Metal Related Activities Prediction by Hidden Markov Models (HMM) Profiles
2.5. Statistical Analysis
3. Results and Discussion
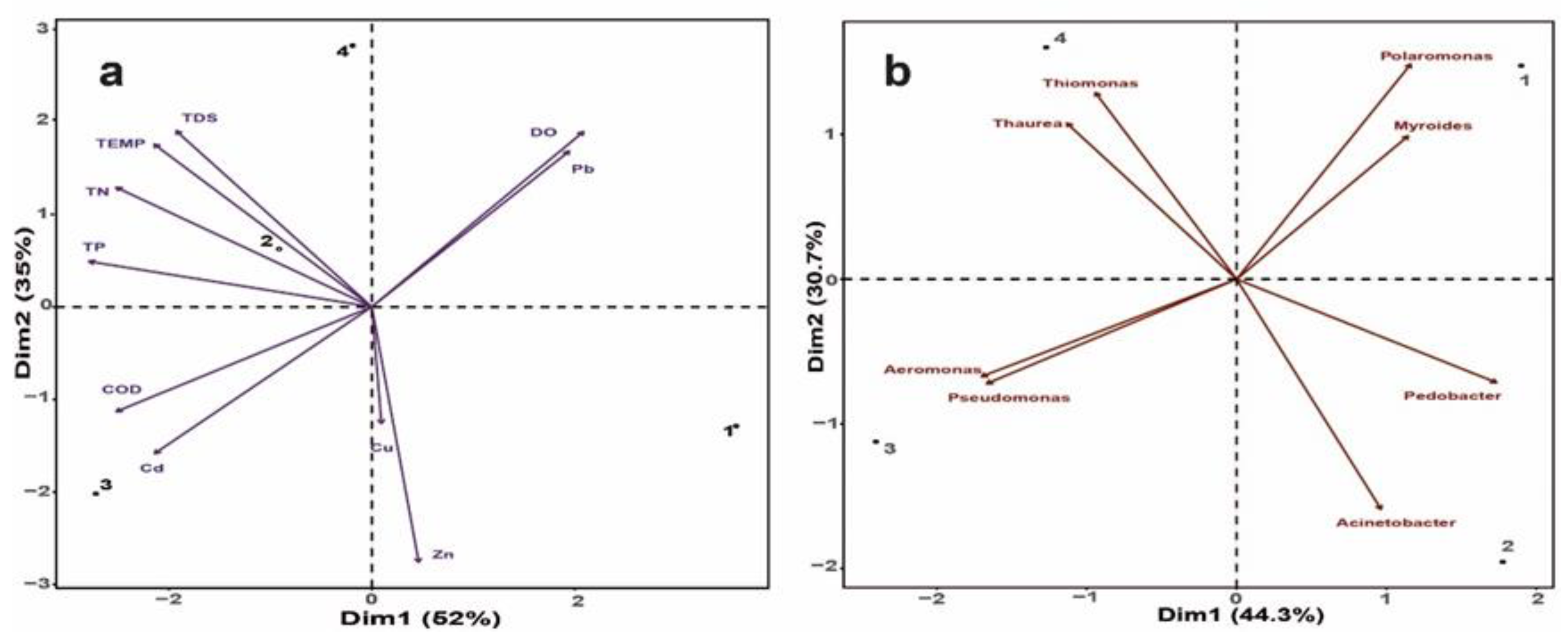
3.1. Pollution and Water Quality in the Apatlaco River Like a Selective Pressure to Autochthonous Microbial Communities
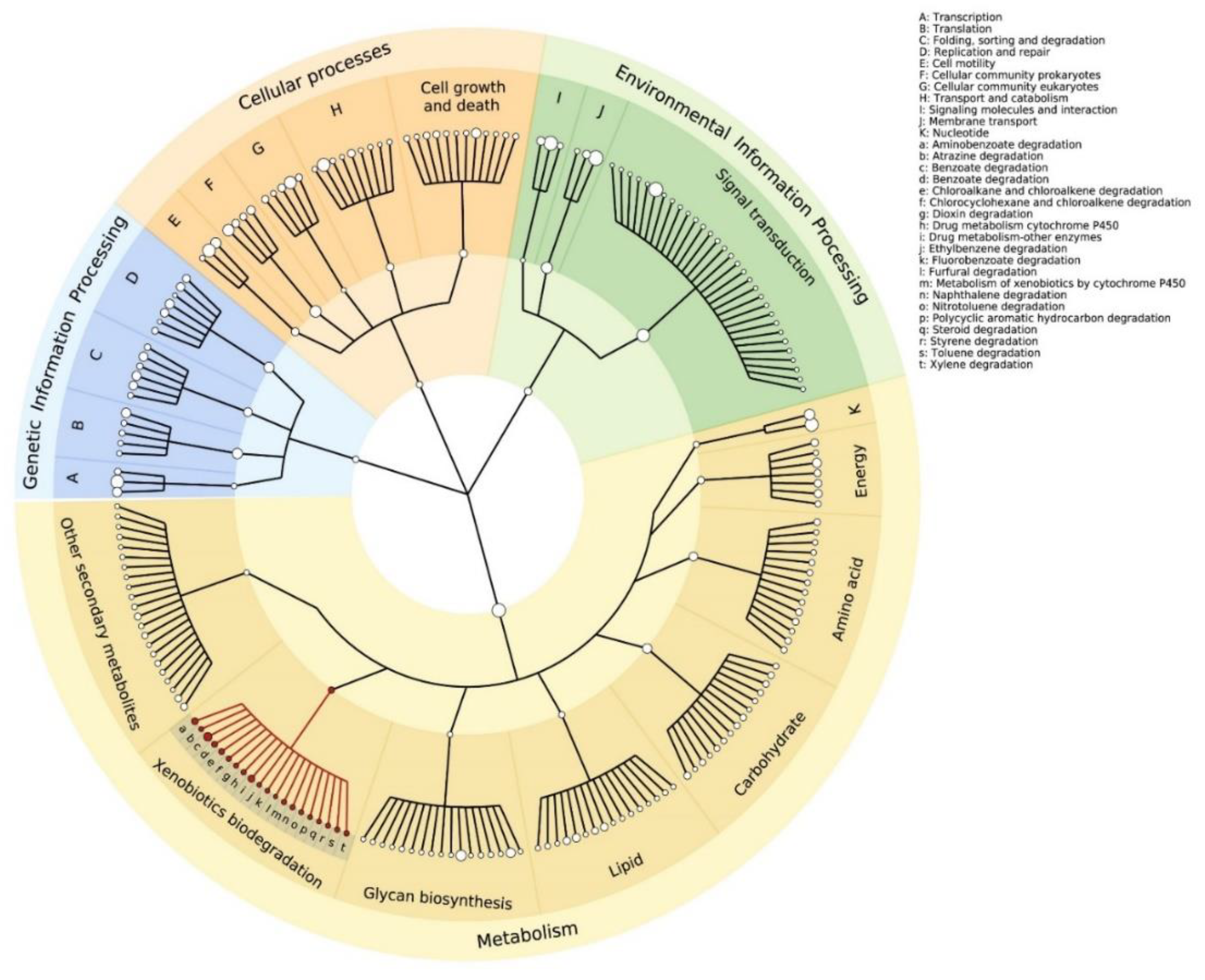
3.2. The Functional Potential of the Microbial Community along the Apatlaco River
3.3. Microbial Genes and Enzymes Involved in the Degradation of Industrial Pollutants: Plastics and Heavy Metals and Metalloids
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, Y.W.; Zhou, L.Q.; Dong, B.Q.; Dai, C. Health assessment for urban rivers based on the pressure, state and response framework—A case study of the Shiwuli River. Ecol. Indic. 2019, 99, 324–331. [Google Scholar] [CrossRef]
- Peters, M.; Guo, Q.; Strauss, H.; Wei, R.; Li, S.; Yue, F. Contamination patterns in river water from rural Beijing: A hydrochemical and multiple stable isotope study. Sci. Total Environ. 2019, 654, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bai, Y.; Guo, Q.; Li, Z.; Qi, M.; Ma, X.; Wang, H.; Kong, D.; Wang, A.; Liang, B. Bioremediation of contaminated urban river sediment with methanol stimulation: Metabolic processes accompanied with microbial community changes. Sci. Total Environ. 2019, 653, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, S.; Zhang, G.; Ren, J.; Zhang, J. Biogeochemistry of nutrients in an estuary affected by human activities: The Wanquan River estuary, eastern Hainan Island, China. Cont. Shelf Res. 2013, 57, 18–31. [Google Scholar] [CrossRef]
- Arora, P.K.; Bae, H. Integration of bioinformatics to biodegradation. Biol. Proced. Online 2014, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Suthersan, S.S. In Situ bioremediation. In Remediation Engineering: Desing Concepts; CRC Press: Boca Raton, FL, USA, 1999; pp. 123–158. ISBN 978-1-56670-137-2. [Google Scholar]
- Ghosh, A.; Bhadury, P. Methods of Assessment of Microbial Diversity in Natural Environments. In Microbial Diversity in the Genomic Era; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Liu, J.; Chen, X.; Shu, H.Y.; Lin, X.R.; Zhou, Q.X.; Bramryd, T.; Shu, W.S.; Huang, L.N. Microbial community structure and function in sediments from e-waste contaminated rivers at Guiyu area of China. Environ. Pollut. 2018, 235, 171–179. [Google Scholar] [CrossRef]
- Megharaj, M.; Ramakrishnan, B.; Venkateswarlu, K.; Sethunathan, N.; Naidu, R. Bioremediation approaches for organic pollutants: A critical perspective. Environ. Int. 2011, 37, 1362–1375. [Google Scholar] [CrossRef]
- González-Toril, E.; Aguilera, Á. Microbial Ecology in Extreme Acidic Environments. In Microbial Diversity in the Genomic Era; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 227–238. [Google Scholar] [CrossRef]
- Techtmann, S.M.; Hazen, C. Metagenomic applications in environmental monitoring and bioremediation. J. Ind. Microbiol. Biotechnol. 2016. [Google Scholar] [CrossRef] [Green Version]
- IMTA. Plan estratégico para la recuperación ambiental de la cuenca del río Apatlaco; IMTA: Jiutepec, Mexico, 2007. [Google Scholar]
- Breton-Deval, L.; Sanchez-Flores, A.; Juárez, K.; Vera-Estrella, R. Integrative study of microbial community dynamics and water quality along The Apatlaco River. Environ. Pollut. 2019, 255, 113158. [Google Scholar] [CrossRef]
- Moeller-Chávez, G.; Seguí-Amórtequi, L.; Alfranca-Burriel, O.; Escalante-Estrada, V.; Pozo-Román, F.; Rivas-Hernández, A. Water reuse in the Apatlaco River Basin (Mexico): A feasibility study. Water Sci. Technol. 2004, 50, 329–337. [Google Scholar] [CrossRef]
- 15. 2540 SOLIDS (2017). Standard Methods For the Examination of Water and Wastewater; American Public Health Association, American Water Works Association, Water Environment Federation: Denver, CO, USA, 2018. [Google Scholar] [CrossRef]
- Andrews. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 14 February 2020).
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, R.; Liu, C.; Leung, C.; Ting, H. MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler driven by Advanced Methodologies and Community Practices. Methods 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hébert, F.O.; Grambauer, S.; Barber, I.; Landry, C.R.; Aubin-Horth, N. Transcriptome sequences spanning key developmental states as a resource for the study of the cestode Schistocephalus solidus, a threespine stickleback parasite. GigaScience 2016, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danso, Z. Supplemental Figures New Insights into the Function and Global Distribution of Polyethylene Terephthalate (PET)-Degrading Bacteria and. Appl. Environ. Microbiol. 2018, 53, 1689–1699. [Google Scholar] [CrossRef]
- Mattes, T.E.; Alexander, A.K.; Richardson, P.M.; Munk, A.C.; Han, C.S.; Stothard, P.; Coleman, N.V. The genome of Polaromonas sp. strain JS666: Insights into the evolution of a hydrocarbon- and xenobiotic-degrading bacterium, and features of relevance to biotechnology. Appl. Environ. Microbiol. 2008, 74, 6405–6416. [Google Scholar] [CrossRef] [Green Version]
- Mekuto, L.; Ntwampe, S.K.O.; Mudumbi, J.B.N.; Akinpelu, E.A.; Mewa-Ngongang, M. Metagenomic data of free cyanide and thiocyanate degrading bacterial communities. Data Brief. 2017, 13, 738–741. [Google Scholar] [CrossRef]
- Naz, T.; Khan, M.D.; Ahmed, I.; Rehman, S.U.; Rha, E.S.; Malook, I.; Jamil, M. Biosorption of heavy metals by Pseudomonas species isolated from sugar industry. Toxicol. Ind. Health 2016, 32, 1619–1627. [Google Scholar] [CrossRef]
- Sinbuathong, N.; Sirirote, P.; Watts, D.; Chulalaksananukul, S. Heavy metal resistant anaerobic bacterial strains from brewery digester sludge. Int. J. Glob. Warm. 2013, 5, 127. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Li, X.; Wang, H.; Li, W. Spatio-temporal variation analysis of hydrochemical characteristics in the Luanhe River Basin, China. Water Sci. Technol. 2013, 67, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, G.; Liu, H.; Lam, P.K.S. Multivariate statistical evaluation of dissolved trace elements and a water quality assessment in the middle reaches of Huaihe River, Anhui, China. Sci. Total Environ. 2017, 583, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Meybeck, M.; Kuusisto, E.; Mäkelä, A.; Mälkki, E. Chapter 2 Water Quality. In Water Quality Assessments—A Guide to Use of Biota, Sediments and Water in Environmental Monitoring; Cambridge University Press: Cambridge, UK, 1996; p. 29. [Google Scholar]
- Tian, S.; Wang, Z.; Shang, H. Study on the self-purification of Juma River. Procedia Environ. Sci. 2011, 11, 1328–1333. [Google Scholar] [CrossRef] [Green Version]
- Atashgahi, S.; Aydin, R.; Dimitrov, M.R.; Sipkema, D.; Hamonts, K.; Lahti, L.; Maphosa, F.; Kruse, T.; Saccenti, E.; Springael, D.; et al. Impact of a wastewater treatment plant on microbial community composition and function in a hyporheic zone of a eutrophic river. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef]
- Bao, Y.; Guo, C.; Wang, H.; Lu, G.; Yang, C.; Chen, M.; Dang, Z. Fe- and S-Metabolizing Microbial Communities Dominate an AMD-Contaminated River Ecosystem and Play Important Roles in Fe and S Cycling. Geomicrobiol. J. 2017, 34, 695–705. [Google Scholar] [CrossRef]
- Zheng, Y.; Xiao, Y.; Yang, Z.H.; Wu, S.; Xu, H.J.; Liang, F.Y.; Zhao, F. The bacterial communities of bioelectrochemical systems associated with the sulfate removal under different pHs. Process Biochem. 2014, 49, 1345–1351. [Google Scholar] [CrossRef]
- Kotresha, D.; Vidyasagar, G.M. Phenol degradation in a packed bed reactor by immobilized cells of Pseudomonas aeruginosa MTCC 4997. Biocatal. Agric. Biotechnol. 2017, 10, 386–389. [Google Scholar] [CrossRef]
- Wasi, S.; Tabrez, S.; Ahmad, M. Use of Pseudomonas spp. for the bioremediation of environmental pollutants: A review. Environ. Monit. Assess. 2013, 185, 8147–8155. [Google Scholar] [CrossRef]
- Hu, S.; Yuan, S.; Qu, H.; Jiang, T.; Zhou, Y.; Wang, M.; Ming, D. Antibiotic resistance mechanisms of Myroides sp. J. Zhejiang Univ. Sci. B 2016, 17, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Kotik, M.; Davidová, A.; Voříšková, J.; Baldrian, P. Bacterial communities in tetrachloroethene-polluted groundwaters: A case study. Sci. Total Environ. 2013, 454–455, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Michaud, L.; Caruso, C.; Mangano, S.; Interdonato, F.; Bruni, V.; Lo Giudice, A. Predominance of Flavobacterium, Pseudomonas, and Polaromonas within the prokaryotic community of freshwater shallow lakes in the northern Victoria Land, East Antarctica. FEMS Microbiol. Ecol. 2012, 82, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; He, S.; Liu, X.; Hu, J. Biobegradation and metabolic mechanism of cyprodinil by strain Acinetobacter sp. from a contaminated-agricultural soil in China. Ecotoxicol. Environ. Saf. 2018, 159, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Paisio, C.E.; Talano, M.A.; González, P.S.; Magallanes-Noguera, C.; Kurina-Sanz, M.; Agostini, E. Biotechnological tools to improve bioremediation of phenol by Acinetobacter sp. RTE1.4. Environ. Technol. 2016, 37, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Qu, B.; Liu, H.; Ding, J.; Ren, N. Analysis of organochlorine pesticides in surface water of the Songhua River using magnetoliposomes as adsorbents coupled with GC-MS/MS detection. Sci. Total Environ. 2018, 618, 70–79. [Google Scholar] [CrossRef]
- Fang, H.; Zhang, H.; Han, L.; Mei, J.; Ge, Q.; Long, Z.; Yu, Y. Exploring bacterial communities and biodegradation genes in activated sludge from pesticide wastewater treatment plants via metagenomic analysis. Environ. Pollut. 2018, 243, 1206–1216. [Google Scholar] [CrossRef]
- Yagi, J.M.; Sims, D.; Brettin, T.; Bruce, D.; Madsen, E.L. The genome of Polaromonas naphthalenivorans strain CJ2, isolated from coal tar-contaminated sediment, reveals physiological and metabolic versatility and evolution through extensive horizontal gene transfer. Environ. Microbiol. 2009, 11, 2253–2270. [Google Scholar] [CrossRef]
- Osborne, T.H.; Jamieson, H.E.; Hudson-Edwards, K.A.; Nordstrom, D.K.; Walker, S.R.; Ward, S.A.; Santini, J.M. Microbial oxidation of arsenite in a subarctic environment: Diversity of arsenite oxidase genes and identification of a psychrotolerant arsenite oxidiser. BMC Microbiol. 2010, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Paul, D.; Jain, R.K. Biofilms: Implications in bioremediation. Trends Microbiol. 2006, 14, 389–397. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Xia, D.; Jiang, X.; Fu, D.; Shen, L.; Wang, H.; Li, Q.B. Enhanced bioreduction of iron and arsenic in sediment by biochar amendment influencing microbial community composition and dissolved organic matter content and composition. J. Hazard. Mater. 2016, 311, 20–29. [Google Scholar] [CrossRef]
- Samal, K.; Dash, R.R.; Bhunia, P. Treatment of wastewater by vermifiltration integrated with macrophyte filter: A review. J. Environ. Chem. Eng. 2017, 5, 2274–2289. [Google Scholar] [CrossRef]
- Roy, U.; Sengupta, S.; Banerjee, P.; Das, P.; Bhowal, A.; Datta, S. Assessment on the decolourization of textile dye (Reactive Yellow) using Pseudomonas sp. immobilized on fly ash: Response surface methodology optimization and toxicity evaluation. J. Environ. Manag. 2018, 223, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, K.; Megharaj, M.; Venkateswarlu, K.; Naidu, R. Bioavailability of weathered hydrocarbons in engine oil-contaminated soil: Impact of bioaugmentation mediated by Pseudomonas spp. on bioremediation. Sci. Total Environ. 2018, 636, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gedalanga, P.B.; Mahendra, S. Advances in bioremediation of 1,4-dioxane-contaminated waters. J. Environ. Manag. 2017, 204, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Fernández, P.M.; Viñarta, S.C.; Bernal, A.R.; Cruz, E.L.; Figueroa, L.I.C. Bioremediation strategies for chromium removal: Current research, scale-up approach and future perspectives. Chemosphere 2018, 208, 139–148. [Google Scholar] [CrossRef]
- Wang, X.; Xue, L.; Chang, S.; He, X.; Fan, T.; Wu, J.; Niu, J.; Emaneghemi, B. Bioremediation and metabolism of clothianidin by mixed bacterial consortia enriched from contaminated soils in Chinese greenhouse. Process Biochem. 2019, 78, 114–122. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Y.; Qin, Z.; Luo, P.; Ma, Z.; Tan, M.; Kang, H.; Huang, Z. A novel manganese oxidizing bacterium-Aeromonas hydrophila strain DS02: Mn(II) oxidization and biogenic Mn oxides generation. J. Hazard. Mater. 2019, 367, 539–545. [Google Scholar] [CrossRef]
- Kumar, S.S.; Shantkriti, S.; Muruganandham, T.; Murugesh, E.; Rane, N.; Govindwar, S.P. Bioinformatics aided microbial approach for bioremediation of wastewater containing textile dyes. Ecol. Inform. 2016, 31, 112–121. [Google Scholar] [CrossRef]
- Uhrynowski, W.; Debiec, K.; Sklodowska, A.; Drewniak, L. The role of dissimilatory arsenate reducing bacteria in the biogeochemical cycle of arsenic based on the physiological and functional analysis of Aeromonas sp. O23A. Sci. Total Environ. 2017, 598, 680–689. [Google Scholar] [CrossRef]
- Du, L.N.; Li, G.; Zhao, Y.H.; Xu, H.K.; Wang, Y.; Zhou, Y.; Wang, L. Efficient metabolism of the azo dye methyl orange by Aeromonas sp. strain DH-6: Characteristics and partial mechanism. Int. Biodeterior. Biodegrad. 2015, 105, 66–72. [Google Scholar] [CrossRef]
- Chen, B.Y.; Hung, J.Y.; Shiau, T.J.; Wei, Y.H. Exploring two-stage fermentation strategy of polyhydroxyalkanoate production using Aeromonas hydrophila. Biochem. Eng. J. 2013, 78, 80–84. [Google Scholar] [CrossRef]
- Argiroff, W.A.; Zak, D.R.; Lanser, C.M.; Wiley, M.J. Microbial Community Functional Potential and Composition Are Shaped by Hydrologic Connectivity in Riverine Floodplain Soils. Microb. Ecol. 2017, 73, 630–644. [Google Scholar] [CrossRef] [PubMed]
- SEMARNAT. Secretaría de Medio Ambiente y Recursos Naturales. nforme de la Situación del Medio Ambiente en México. Compendio de Estadísticas Ambientales. Indicadores Clave, de Desempeño Ambiental y de Crecimiento Verde; SEMARNAT: Ciudad de México, México, 2016; p. 380. [Google Scholar]
- Ho, B.T.; Roberts, T.K.; Lucas, S. An overview on biodegradation of polystyrene and modified polystyrene: The microbial approach. Crit. Rev. Biotechnol. 2018, 38, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Otto, K.; Hofstetter, K.; Ro, M.; Witholt, B.; Schmid, A. Biochemical Characterization of StyAB from. J. Bacteriol. 2004, 186, 5292–5302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischler, D.; Eulberg, D.; Lakner, S.; Kaschabek, S.R.; Van Berkel, W.J.H.; Schlömann, M. Identification of a novel self-sufficient styrene monooxygenase from Rhodococcus opacus 1CP. J. Bacteriol. 2009, 191, 4996–5009. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.L.; Liu, W.; Tang, C.; Franks, A.E. Microorganisms in heavy metal bioremediation: Strategies for applying microbial-community engineering to remediate soils. AIMS Bioeng. 2016, 3, 211–229. [Google Scholar] [CrossRef]
- Sharma, V.; Pant, D. Structural basis for expanding the application of bioligand in metal bioremediation: A review. Bioresour. Technol. 2018, 252, 188–197. [Google Scholar] [CrossRef]
- Gutiérrez, J.-C.; de Francisco, P.; Amaro, F.; Díaz, S.; Martín-González, A. Structural and Functional Diversity of Microbial Metallothionein Genes. In Microbial Diversity in the Genomic Era; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 387–407. [Google Scholar] [CrossRef]
- Marzan, L.W.; Hossain, M.; Mina, S.A.; Akter, Y.; Chowdhury, A.M.M.A. Isolation and biochemical characterization of heavy-metal resistant bacteria from tannery effluent in Chittagong city, Bangladesh: Bioremediation viewpoint. Egypt. J. Aquat. Res. 2017, 43, 65–74. [Google Scholar] [CrossRef]
- Argudín, M.A.; Hoefer, A.; Butaye, P. Heavy metal resistance in bacteria from animals. Res. Vet. Sci. 2019, 122, 132–147. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pollutants | S1 | S2 | S3 | S4 |
|---|---|---|---|---|
| COD (mg/L) | 80 ± 9.36 | 270 ± 23. 35 | 480 ± 31 | 110 ± 16 |
| TN (mg/L) | 6 ± 1.48 | 31 ± 6.74 | 38 ± 5.78 | 45 ± 6.45 |
| DO (mg/L) | 7 ± 0.59 | 4 ± 0.38 | 2 ± 1.19 | 7 ± 1.98 |
| TP (mg/L) | 0.77 ± 0.22 | 9 ± 2.65 | 16 ± 2.45 | 9 ± 1.47 |
| TDS (mg/L) | 62 ± 1.77 | 206 ± 9.75 | 299 ± 10.35 | 756 ± 20.67 |
| Temp (°C) | 17 ± 1.56 | 22 ± 1.10 | 23 ± 1.21 | 26 ± 2.25 |
| Zn (mg/L) | 0.20 ± 0.02 | 0.16 ± 0.02 | 0.20 ± 0.02 | 0.13 ± 0.01 |
| Cd (mg/L) | 0.07 ± 0.02 | 0.16 ± 0.01 | 0.19 ± 0.01 | 0.16 ± 0.01 |
| Pb (mg/L) | 3.95 ± 0.22 | 2.53 ± 0.27 | 2.24 ± 0.08 | 4.35 ± 0.15 |
| Cu (mg/L) | 0.99 ± 0.04 | 0.46 ± 0.02 | 1.15 ± 0.06 | 0.88 ± 0.03 |
| Mn (mg/L) | <0.0015 | <0.0015 | <0.0015 | <0.0015 |
| Cr (mg/L) | <0.003 | <0.003 | <0.003 | <0.003 |
| Parameter | S1 | S2 | S3 | S4 |
|---|---|---|---|---|
| Total filtered pair-end reads | 41771406 | 64325748 | 64321034 | 47153792 |
| Assembly size (Mb) | 42.716 | 82.952 | 78.809 | 48.360 |
| Total contigs | 45488 | 103533 | 114279 | 59123 |
| N50/L50 | 1959/4015 | 1626/9243 | 1165/14 | 1694/5245 |
| Total predicted genes | 75,951 | 164,571 | 165,377 | 93,227 |
| Total annotated genes | 26,910 | 35,597 | 44,547 | 28,939 |
| Total tax. Assig. (genus) | 73 | 108 | 140 | 136 |
| Microorganism | Rel. Abundance (%) | Compounds | Pathogen Ref. | ||||
|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | ||||
| Thiomonas sp. | 1.17 | 0.75 | 1.30 | 4.63 | As, S, | No | [6,7,8,34] |
| Polaromonas sp. | 1.74 | 0.41 | 0.24 | 0.73 | Hg, As, alkanes, Pyrene | No | [23,44,45,46] |
| Pedobacter sp. | 1.33 | 2.46 | 0.24 | 0.80 | As(V) | opportunistic | [47] |
| Myroides sp. | 27.2 | 5.19 | 6.20 | 4.03 | CN−, SCN− Organic matter | opportunistic | [24,48] |
| Pseudomonas sp. | 0.72 | 1.77 | 5.07 | 7.58 | Pb +2, Ni +2, Cu +2, Cr +3, NO3, detergents, dyes, pesticides, hydrocarbons | opportunistic | [25,36,49,50] |
| Acinetobacter sp. | 0.30 | 50.2 | 5.79 | 0.72 | Hydroxydioxane, Cr, Clothianidin, cyprodinil, | yes | [40,51,52,53] |
| Aeromonas sp. | 0.29 | 0.25 | 2.03 | 0.54 | As, Cu, Fe, Ni, Zn, Mn (II), dyes, PHAs, | yes | [54,55,56,57,58] |
| Tavera sp. | 0.16 | 0.15 | 0.36 | 1.29 | Zn, Cd, Co, Cu, Ni, Pb, Cr, Hg, Se | No | [43] |
| DEGRADATION PATHWAYS GENE_ID | Site | Target Compound | Enzymes | Taxonomic Assignation |
|---|---|---|---|---|
| MT023458 | S1 | PET | S9 family peptidase | Flavobacteriia bacterium |
| MT023459 | S1 | |||
| MT023460 | S3 | OsmC family protein Esterase | Proteobacteria | |
| MT023461 | S1 | Alpha/beta hydrolase | Bordetella flabilis | |
| MT023462 | S2 | dipeptidyl aminopeptidase | Sphingobacteriales bacterium | |
| MT023463 | S4 | alpha/beta hydrolase | Bacteroidia bacterium | |
| MT023464 | S2 | |||
| MT023465 | S4 | |||
| MT023466 | S3 | |||
| MT023467 | S3 | alpha/beta hydrolase | Lutibacter sp. | |
| MT023468 | S1 | alpha/beta hydrolase | Flavobacterium aquicola | |
| MT023469 | S2 | alpha/beta hydrolase | Flavobacterium aquicola | |
| MT023470 | S3 | alpha/beta hydrolase | Flavobacterium aquicola | |
| MT023471 | S1 | Polystyrene | kynurenine 3-monooxygenase | Flavobacteria bacterium |
| MT023472 | S1 | FAD-dependent oxidoreductase | Chitinophagaceae bacterium | |
| MT023473 | S2 | Ferredoxin | Aurantimicrobium sp. | |
| MT023474 | S2 | FAD-dependent monooxygenase | Flavobacteriia bacterium | |
| MT023475 | S2 | Ubiquinone biosynthesis protein UbiH | Coxiellaceae bacterium | |
| MT023476 | S4 | Ferredoxin | Aurantimicrobium sp. |
| Degradation Pathway Gene_ID | Site | Target Compound | Taxonomic Assignation |
|---|---|---|---|
| MT023477 | S1 | Cd | Flavobacterium branchiophilum |
| MT023478 | S1 | Methylotenera versatilis | |
| MT023479 | S1 | Methylotenera versatilis | |
| MT023480 | S1 | Emticiciaoligo trophica | |
| MT023481 | S1 | Azospirillum sp. B510 | |
| MT023482 | S1 | Dechloromonas aromatica | |
| MT023483 | S1 | Sphingopyxis sp. QXT-31 | |
| MT023484 | S1 | Flavobacterium branchiophilum | |
| MT023485 | S2 | Methylotenera mobilis | |
| MT023486 | S2 | Chryseobacterium taklimakanense | |
| MT023487 | S2 | Polynucleobacter duraquae | |
| MT023488 | S2 | Cytophaga hutchinsonii | |
| MT023489 | S2 | Acinetobacter schindleri | |
| MT023490 | S2 | Myroides odoratimimus | |
| MT023491 | S2 | Acinetobacter schindleri | |
| MT023492 | S3 | Acidovorax citrulli | |
| MT023493 | S3 | Flavobacteriuman huiense | |
| MT023494 | S3 | Flavobacterium commune | |
| MT023495 | S3 | Flavobacterium columnare | |
| MT023496 | S3 | Chryseobacterium taklimakanense | |
| MT023497 | S3 | Azospira oryzae | |
| MT023498 | S3 | Polynucleobacter duraquae | |
| MT023499 | S3 | Myroides sp. A21 | |
| MT023500 | S3 | Azospira oryzae | |
| MT023501 | S3 | Flavobacterium commune | |
| MT023502 | S3 | Sulfurimonas autotrophica | |
| MT023503 | S4 | Methylotenera mobilis | |
| MT023504 | S4 | Runella slithyformis | |
| MT023505 | S4 | Polynucleobacter duraquae | |
| MT023506 | S4 | Acidovorax citrulli | |
| MT023507 | S4 | Shewanella sp. ANA-3 | |
| MT023437 | S1 | Pb | Flavobacterium johnsoniae UW101 |
| MT023438 | S1 | Limnohabitans sp. 63ED37-2 | |
| MT023439 | S1, S2 | Limnohabitans sp. 103DPR2 | |
| MT023440 | S1, S2 | Limnohabitans sp. 103DPR2 | |
| MT023441 | S2 | Azoarcus olearius | |
| MT023442 | S2 | Polynucleobacter asymbioticus | |
| MT023443 | S2 | Acinetobacter johnsonii | |
| MT023444 | S2 | Acinetobacter schindleri | |
| MT023445 | S2 | Azoarcus olearius | |
| MT023446 | S3 | Acidovorax sp. 1608163 | |
| MT023447 | S3 | Alicycliphilus denitrificans BC | |
| MT023448 | S3 | Alicycliphilus denitrificans BC | |
| MT023449 | S3 | Chryseobacterium taklimakanense | |
| MT023450 | S3 | Acinetobacter johnsonii | |
| MT023451 | S4 | Cellvibrio sp. PSBB006 | |
| MT023452 | S4 | Polynucleobacter asymbioticus | |
| MT023453 | S4 | Limnohabitan ssp. 63ED37-2 | |
| MT023454 | S4 | Beta proteobacterium CB | |
| MT023455 | S4 | Limnohabitan ssp. 63ED37-2 | |
| MT023456 | S4 | Azoarcus olearius | |
| MT023457 | S4 | Acinetobacter johnsonii |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breton-Deval, L.; Sanchez-Reyes, A.; Sanchez-Flores, A.; Juárez, K.; Salinas-Peralta, I.; Mussali-Galante, P. Functional Analysis of a Polluted River Microbiome Reveals a Metabolic Potential for Bioremediation. Microorganisms 2020, 8, 554. https://doi.org/10.3390/microorganisms8040554
Breton-Deval L, Sanchez-Reyes A, Sanchez-Flores A, Juárez K, Salinas-Peralta I, Mussali-Galante P. Functional Analysis of a Polluted River Microbiome Reveals a Metabolic Potential for Bioremediation. Microorganisms. 2020; 8(4):554. https://doi.org/10.3390/microorganisms8040554
Chicago/Turabian StyleBreton-Deval, Luz, Ayixon Sanchez-Reyes, Alejandro Sanchez-Flores, Katy Juárez, Ilse Salinas-Peralta, and Patricia Mussali-Galante. 2020. "Functional Analysis of a Polluted River Microbiome Reveals a Metabolic Potential for Bioremediation" Microorganisms 8, no. 4: 554. https://doi.org/10.3390/microorganisms8040554