Soil Bacterial Diversity and Potential Functions Are Regulated by Long-Term Conservation Tillage and Straw Mulching

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description
2.2. Experimental Design
2.3. Soil Sampling and Physicochemical Properties Analysis
2.4. DNA Extraction, PCR Amplification, and Illumina Sequencing
2.5. Data Processing and Bioinformatics Analysis
2.6. Statistical Analysis
3. Results
3.1. Soil physicochemical Properties
3.2. Sequencing Depth and Alpha Diversity
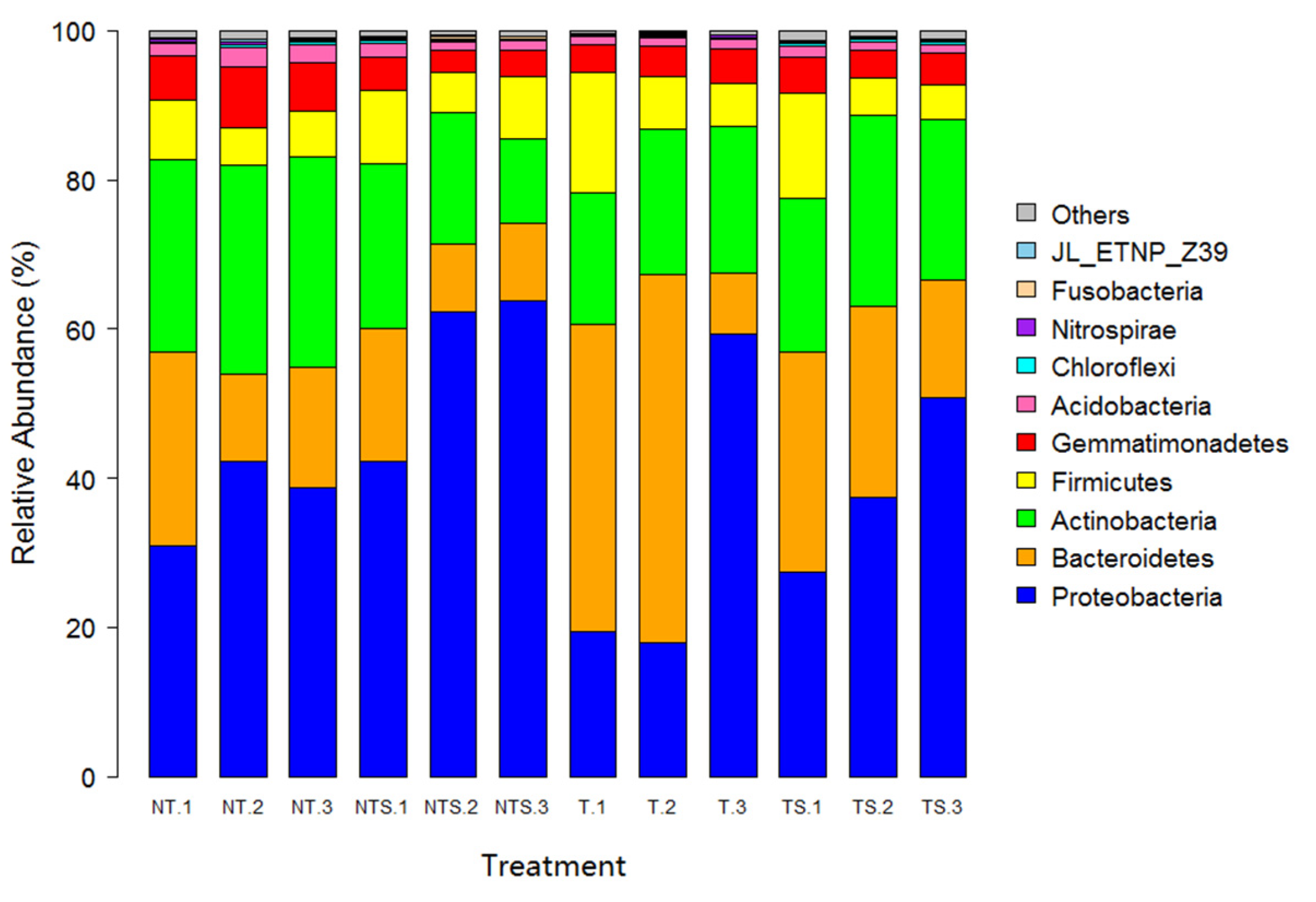
3.3. Beta Diversity and Microbial Community Composition
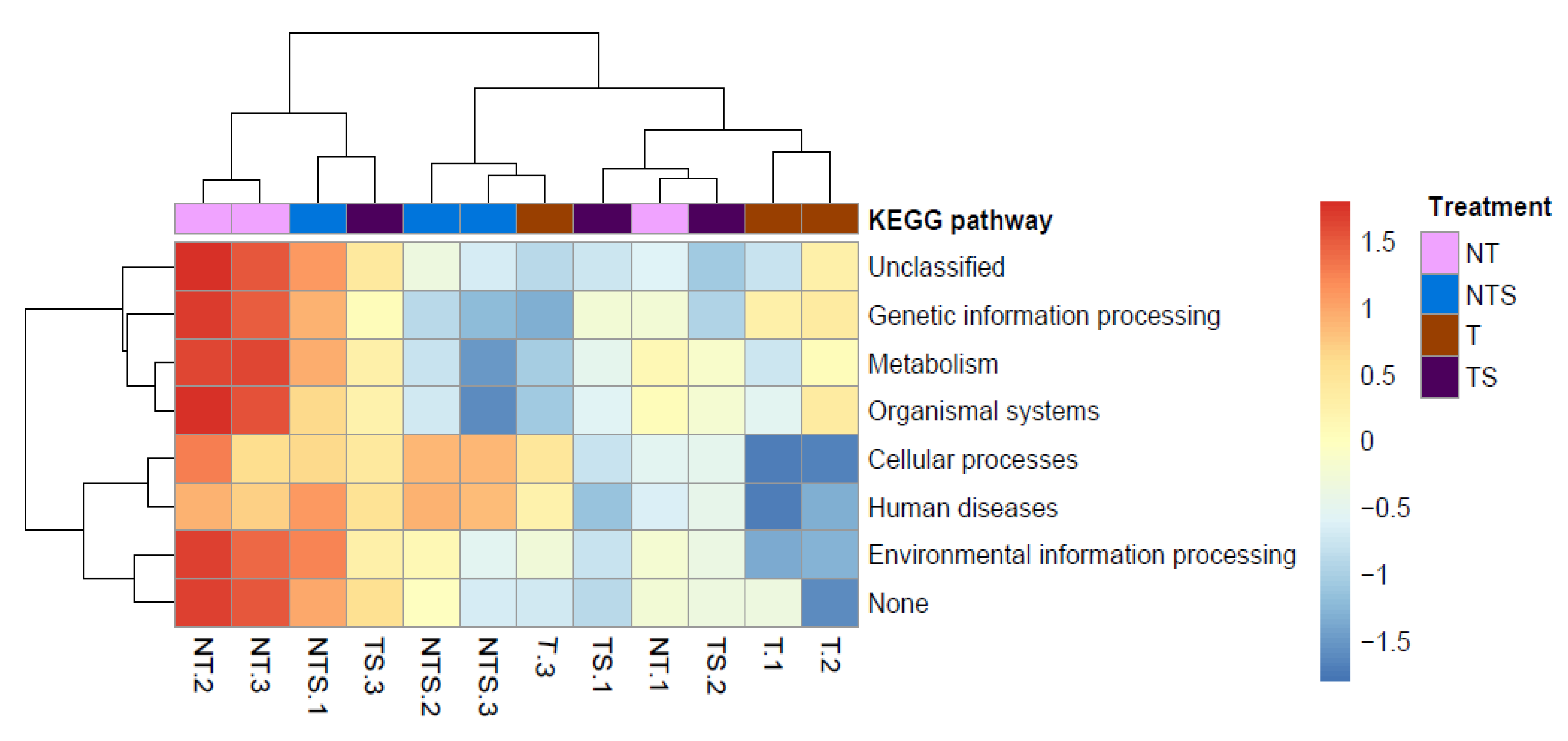
3.4. Variation of Predicted Functions of Soil Microbial Community
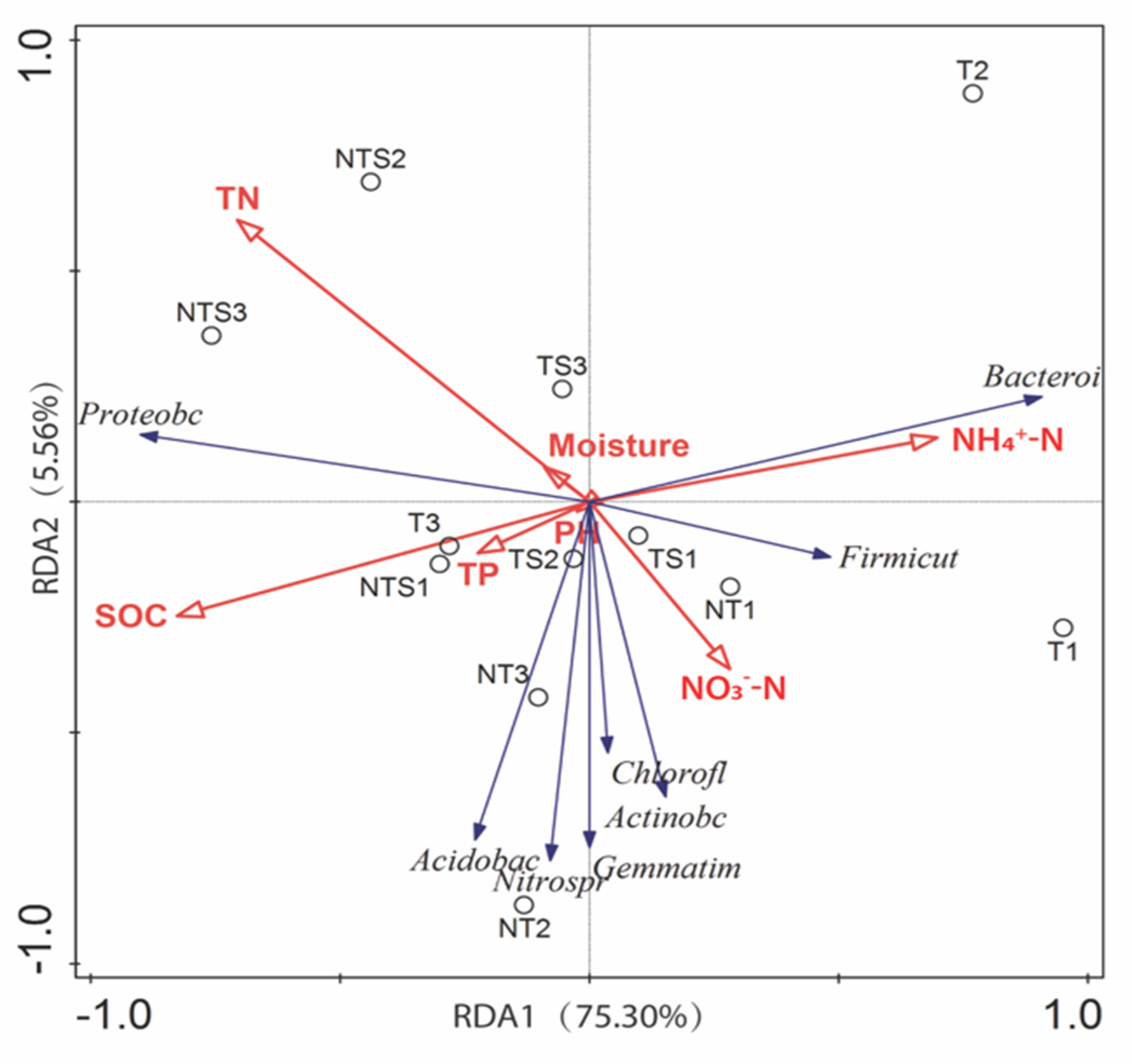
3.5. Relationships between Soil Parameters and Microbial Community Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Congreves, K.; Hayes, A.; Verhallen, E.; Van Eerd, L. Long-term impact of tillage and crop rotation on soil health at four temperate agroecosystems. Soil Tillage Res. 2015, 152, 17–28. [Google Scholar] [CrossRef]
- Morrow, J.G.; Huggins, D.R.; Carpenter-Boggs, L.A.; Reganold, J.P. Evaluating measures to assess soil health in long-term agroecosystem trials. Soil Sci. Soc. Am. J. 2016, 80, 450–462. [Google Scholar] [CrossRef]
- Al-Kaisi, M.M.; Lowery, B. Soil Health and Intensification of Agroecosystems; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Karlen, D.L.; Veum, K.S.; Sudduth, K.A.; Obrycki, J.F.; Nunes, M.R. Soil health assessment: Past accomplishments, current activities, and future opportunities. Soil Tillage Res. 2019, 195, 104365. [Google Scholar] [CrossRef]
- Lavecchia, A.; Curci, M.; Jangid, K.; Whitman, W.B.; Ricciuti, P.; Pascazio, S.; Crecchio, C. Microbial 16S gene-based composition of a sorghum cropped rhizosphere soil under different fertilization managements. Biol. Fertil. Soils 2015, 51, 661–672. [Google Scholar] [CrossRef]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Huang, M.; Jiang, L.; Zou, Y.; Xu, S.; Deng, G. Changes in soil microbial properties with no-tillage in Chinese cropping systems. Biol. Fertil. Soils 2013, 49, 373–377. [Google Scholar] [CrossRef]
- Liu, K.; Blackshaw, R.E.; Johnson, E.N.; Hossain, Z.; Hamel, C.; St-Arnaud, M.; Gan, Y. Lentil enhances the productivity and stability of oilseed-cereal cropping systems across different environments. Eur. J. Agron. 2019, 105, 24–31. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579. [Google Scholar] [CrossRef] [PubMed]
- Gil, S.V.; Meriles, J.; Conforto, C.; Figoni, G.; Basanta, M.; Lovera, E.; March, G.J. Field assessment of soil biological and chemical quality in response to crop management practices. World J. Microbiol. Biotechnol. 2009, 25, 439–448. [Google Scholar]
- Moraru, P.I.; Rusu, T.J. Soil tillage conservation and its effect on soil organic matter, water management and carbon sequestration. J. Food Agric. Environ. 2010, 8, 309–312. [Google Scholar]
- Nannipieri, P.; Ascher, J.; Ceccherini, M.; Landi, L.; Pietramellara, G.; Renella, G. Microbial diversity and soil functions. Eur. J. Soil Sci. 2017, 68, 12–26. [Google Scholar] [CrossRef]
- Beauregard, M.; Hamel, C.; St-Arnaud, M. Long-term phosphorus fertilization impacts soil fungal and bacterial diversity but not AM fungal community in alfalfa. Microb. Ecol. 2010, 59, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hao, Z.; Sun, Y.; Guo, L.; Huang, L.; Zeng, Y.; Wang, Y.; Yang, L.; Chen, B. Comparison on the structure and function of the rhizosphere microbial community between healthy and root-rot Panax notoginseng. Appl. Soil Ecol. 2016, 107, 99–107. [Google Scholar] [CrossRef]
- Yeboah, S.; Zhang, R.; Cai, L.; Li, L.; Xie, J.; Luo, Z.; Liu, J.; Wu, J. Tillage effect on soil organic carbon, microbial biomass carbon and crop yield in spring wheat-field pea rotation. Plant Soil Environ. 2016, 62, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, A.; Dick, W.A. Bacterial community diversity in soil under two tillage practices as determined by pyrosequencing. Microb. Ecol. 2015, 70, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, Y.; Ren, T.; Tian, Z.; Wang, G.; He, X.; Tian, C. Short-term effect of tillage and crop rotation on microbial community structure and enzyme activities of a clay loam soil. Biol. Fertil. Soils 2014, 50, 1077–1085. [Google Scholar] [CrossRef]
- Govaerts, B.; Mezzalama, M.; Unno, Y.; Sayre, K.D.; Luna-Guido, M.; Vanherck, K.; Dendooven, L.; Deckers, J. Influence of tillage, residue management, and crop rotation on soil microbial biomass and catabolic diversity. Appl. Soil Ecol. 2007, 37, 18–30. [Google Scholar] [CrossRef]
- Valboa, G.; Lagomarsino, A.; Brandi, G.; Agnelli, A.; Simoncini, S.; Papini, R.; Vignozzi, N.; Pellegrini, S. Long-term variations in soil organic matter under different tillage intensities. Soil Tillage Res. 2015, 154, 126–135. [Google Scholar] [CrossRef]
- Gajda, A.M.; Czyż, E.A.; Furtak, K.; Jończyk, K. Effects of crop production practices on soil characteristics and metabolic diversity of microbial communities under winter wheat. Soil Res. 2019, 57, 124–131. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, L.; Chen, Q.; Wen, X.; Liao, Y. Conservation tillage increases soil bacterial diversity in the dryland of northern China. Agron. Sustain. Dev. 2016, 36, 28. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef] [PubMed]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S. The soil microbiome influences grapevine-associated microbiota. mBio 2015, 6, e02527-02514. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Dai, T.; Tang, Y.; Tao, Y.; Huang, B.; Mu, Q.; Wen, D. Sediment bacterial community structures and their predicted functions implied the impacts from natural processes and anthropogenic activities in coastal area. Mar. Pollut. Bull. 2018, 131, 481–495. [Google Scholar] [CrossRef]
- Al-hebshi, N.N.; Alharbi, F.A.; Mahri, M.; Chen, T. Differences in the bacteriome of smokeless tobacco products with different oral carcinogenicity: Compositional and predicted functional analysis. Genes 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Hamel, C.; Gan, Y.; Sokolski, S.; Bainard, L.D. High frequency cropping of pulses modifies soil nitrogen level and the rhizosphere bacterial microbiome in 4-year rotation systems of the semiarid prairie. Appl. Soil Ecol. 2018, 126, 47–56. [Google Scholar] [CrossRef]
- Bainard, L.D.; Hamel, C.; Gan, Y. Edaphic properties override the influence of crops on the composition of the soil bacterial community in a semiarid agroecosystem. Appl. Soil Ecol. 2016, 105, 160–168. [Google Scholar] [CrossRef]
- Navarro-Noya, Y.E.; Gómez-Acata, S.; Montoya-Ciriaco, N.; Rojas-Valdez, A.; Suárez-Arriaga, M.C.; Valenzuela-Encinas, C.; Jiménez-Bueno, N.; Verhulst, N.; Govaerts, B.; Dendooven, L. Relative impacts of tillage, residue management and crop-rotation on soil bacterial communities in a semi-arid agroecosystem. Soil Biol. Biochem. 2013, 65, 86–95. [Google Scholar] [CrossRef]
- Dong, W.-Y.; Zhang, X.-Y.; Dai, X.-Q.; Fu, X.-L.; Yang, F.-T.; Liu, X.-Y.; Sun, X.-M.; Wen, X.-F.; Schaeffer, S. Changes in soil microbial community composition in response to fertilization of paddy soils in subtropical China. Appl. Soil Ecol. 2014, 84, 140–147. [Google Scholar] [CrossRef]
- Borrell, A.N.; Shi, Y.; Gan, Y.; Bainard, L.; Germida, J.; Hamel, C. Fungal diversity associated with pulses and its influence on the subsequent wheat crop in the Canadian prairies. Plant Soil 2017, 414, 13–31. [Google Scholar] [CrossRef]
- Niu, Y.; Bainard, L.D.; May, W.E.; Hossain, Z.; Hamel, C.; Gan, Y. Intensified pulse rotations buildup pea rhizosphere pathogens in cereal and pulse based cropping systems. Front. Microbiol. 2018, 9, 1909. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, T.B.; Mazzoncini, M.; Bàrberi, P.; Antichi, D.; Silvestri, N. Fifteen years of no till increase soil organic matter, microbial biomass and arthropod diversity in cover crop-based arable cropping systems. Agron. Sustain. Dev. 2012, 32, 853–863. [Google Scholar] [CrossRef]
- Helgason, B.; Walley, F.; Germida, J. No-till soil management increases microbial biomass and alters community profiles in soil aggregates. Appl. Soil Ecol. 2010, 46, 390–397. [Google Scholar] [CrossRef]
- Balota, E.L.; Colozzi-Filho, A.; Andrade, D.S.; Dick, R.P. Microbial biomass in soils under different tillage and crop rotation systems. Biol. Fertil. Soils 2003, 38, 15–20. [Google Scholar] [CrossRef]
- Liu, C.; Cutforth, H.; Chai, Q.; Gan, Y. Farming tactics to reduce the carbon footprint of crop cultivation in semiarid areas. A review. Agron. Sustain. Dev. 2016, 36, 69. [Google Scholar] [CrossRef] [Green Version]
- Helgason, B.; Walley, F.; Germida, J. Biochemistry. Long-term no-till management affects microbial biomass but not community composition in Canadian prairie agroecosytems. Soil Biol. Biochem. 2010, 42, 2192–2202. [Google Scholar] [CrossRef]
- Kay, B.; VandenBygaart, A. Conservation tillage and depth stratification of porosity and soil organic matter. Soil Tillage Res. 2002, 66, 107–118. [Google Scholar] [CrossRef]
- Guo, L.J.; Zhang, Z.S.; Wang, D.D.; Li, C.F.; Cao, C.G. Effects of short-term conservation management practices on soil organic carbon fractions and microbial community composition under a rice-wheat rotation system. Biol. Fertil. Soils 2015, 51, 65–75. [Google Scholar] [CrossRef]
- Zhu, L.; Hu, N.; Zhang, Z.; Xu, J.; Tao, B.; Meng, Y. Short-term responses of soil organic carbon and carbon pool management index to different annual straw return rates in a rice–wheat cropping system. Catena 2015, 135, 283–289. [Google Scholar] [CrossRef]
- Bainard, L.D.; Navarro-Borrell, A.; Hamel, C.; Braun, K.; Hanson, K.; Gan, Y. Increasing the frequency of pulses in crop rotations reduces soil fungal diversity and increases the proportion of fungal pathotrophs in a semiarid agroecosystem. Agric. Ecosyst. Environ. 2017, 240, 206–214. [Google Scholar] [CrossRef]
- Van der Heijden, M.G.; Wagg, C. Soil microbial diversity and agro-ecosystem functioning. Plant Soil 2013, 363, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Xiao, Y.; Li, L.; Jiang, C.; Zu, C.; Li, T.; Cao, H. Impact of tillage practices on soil bacterial diversity and composition under the tobacco-rice rotation in China. J. Microbiol. 2017, 55, 349–356. [Google Scholar] [CrossRef]
- Ramirez, K.S.; Lauber, C.L.; Knight, R.; Bradford, M.A.; Fierer, N. Consistent effects of nitrogen fertilization on soil bacterial communities in contrasting systems. Ecology 2010, 91, 3463–3470. [Google Scholar] [CrossRef]
- Aslam, Z.; Yasir, M.; Yoon, H.S.; Jeon, C.O.; Chung, Y.R. Diversity of the bacterial community in the rice rhizosphere managed under conventional and no-tillage practices. J. Microbiol. 2013, 51, 747–756. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.N.; Rushton, S.P.; Lanyon, C.V.; Whiteley, A.S.; Waite, I.S.; Brookes, P.C.; Kemmitt, S.; Evershed, R.P.; O’Donnell, A.G. Taxon-specific responses of soil bacteria to the addition of low level C inputs. Soil Biol. Biochem. 2010, 42, 1624–1631. [Google Scholar] [CrossRef]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The ecology of Acidobacteria: Moving beyond genes and genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ye, D.; Wang, X.; Settles, M.L.; Wang, J.; Hao, Z.; Zhou, L.; Dong, P.; Jiang, Y.; Ma, Z.S. Soil bacterial communities of different natural forest types in Northeast China. Plant Soil 2014, 383, 203–216. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; van Elsas, J.D.; Olff, H.; Salles, J.F. Dispersal-competition tradeoff in microbiomes in the quest for land colonization. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, J.; Xiu, Z.; Meng, W.; Zhou, Y.; Jin, X.; Qi, G.; Zhang, Y. Gene diversity and its function in the soil microbiome for moss crusts found southeast of the Tengger Desert. Biodivers. Sci. 2018, 7, 727–737. [Google Scholar] [CrossRef]
- Aislabie, J.; Deslippe, J.R.; Dymond, J. Soil Microbes and Their Contribution to Soil Services. In Ecosystems Services in New Zealand Conditions and Trends; Dymond, J.R., Ed.; Manaaki Press: Lincoln, New Zealand, 2013; pp. 143–161. [Google Scholar]
- Eilers, K.G.; Debenport, S.; Anderson, S.; Fierer, N. Digging deeper to find unique microbial communities: The strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol. Biochem. 2012, 50, 58–65. [Google Scholar] [CrossRef]
- Treseder, K.K.; Kivlin, S.N.; Hawkes, C.V. Evolutionary trade-offs among decomposers determine responses to nitrogen enrichment. Ecol. Lett. 2011, 14, 933–938. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Luo, C.; Jiang, L.; Song, M.; Zhang, D.; Li, J.; Li, Y.; Ostle, N.J.; Zhang, G. Land-use changes alter soil bacterial composition and diversity in tropical forest soil in China. Sci. Total Environ. 2020, 712, 136526. [Google Scholar] [CrossRef]
- Chen, H.; Wang, M.; Chang, S. Disentangling Community Structure of Ecological System in Activated Sludge: Core Communities, Functionality, and Functional Redundancy. Microb. Ecol. 2020. [Google Scholar] [CrossRef]
- Alami, M.M.; Xue, J.; Ma, Y.; Zhu, D.; Gong, Z.; Shu, S.; Wang, X. Structure, diversity, and composition of bacterial communities in rhizospheric soil of coptis chinensis franch under continuously cropped fields. Diversity 2020, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Rathour, R.; Jain, K.; Madamwar, D.; Desai, C. Microaerophilic biodegradation of raw textile effluent by synergistic activity of bacterial community DR4. J. Environ. Manag. 2019, 250, 109549. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L.; Tang, C.; Franks, A.E. Competitive traits are more important than stress-tolerance traits in a cadmium-contaminated rhizosphere: A role for trait theory in microbial ecology. Front. Microbiol. 2018, 9, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, D.; Huang, Z.; Zeng, S.; Liu, J.; Wei, D.; Deng, X.; Weng, S.; He, Z.; He, J. Environmental factors shape water microbial community structure and function in shrimp cultural enclosure ecosystems. Front. Microbiol. 2017, 8, 2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Ye, X.; Zhang, H.; Chen, H.; Zhang, D.; Liu, L. Regional variations in the diversity and predicted metabolic potential of benthic prokaryotes in coastal northern Zhejiang, East China Sea. Sci. Rep. 2016, 6, 38709. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Parameter a | Treatment b | |||
|---|---|---|---|---|
| NT | NTS | T | TS | |
| pH | 8.12 ± 0.10 a c | 8.07 ± 0.08 a | 8.07 ± 0.03 a | 8.02 ± 0.12 a |
| Moisture (%) | 12.21 ± 1.02 a | 12.16 ± 0.33 a | 12.54 ± 1.13 a | 12.03 ± 0.37 a |
| SOC (g kg−1) | 13.07 ± 0.25 ab | 13.20 ± 0.26 a | 12.50 ± 0.53 b | 13.03 ± 0.25 ab |
| TN (g kg−1) | 0.83 ± 0.05 b | 0.96 ± 0.06 a | 0.84 ± 0.08 b | 0.85 ± 0.05 b |
| TP (g kg−1) | 0.77 ± 0.15 a | 0.73 ± 0.07 ab | 0.71 ± 0.14 ab | 0.52 ± 0.13 b |
| NO3–N (mg kg−1) | 38.53 ± 2.21 a | 32.26 ± 2.29 ab | 36.24 ± 2.38 ab | 27.73 ± 9.48 b |
| NH4–N (mg kg−1) | 1.41 ± 0.44 a | 1.13 ± 0.34 a | 1.49 ± 0.50 a | 1.37 ± 0.23 a |
| KEGG Pathway | Treatment a | |||
|---|---|---|---|---|
| NT | NTS | T | TS | |
| Environmental information processing | 13.74 ± 0.06 a b | 14.49 ± 0.36 a | 13.44 ± 1.12 a | 13.78 ± 0.17 a |
| Genetic information processing | 15.58 ± 0.06 a | 15.59 ± 0.23 a | 16.41 ± 0.99 a | 15.68 ± 0.60 a |
| Metabolism | 52.28 ± 0.36 a | 50.09 ± 1.45 b | 51.40 ± 1.20 ab | 51.97 ± 0.66 ab |
| Organismal systems | 0.862 ± 0.003 a | 0.817 ± 0.018 b | 0.863 ± 0.036 a | 0.852 ± 0.010 ab |
| Cellular processes | 3.51 ± 0.21 a | 4.16 ± 0.47 a | 3.24 ± 0.82 a | 3.51 ± 0.20 a |
| Human diseases | 0.92 ± 0.02 b | 1.11 ± 0.09 a | 0.91 ± 0.15 b | 0.94 ± 0.07 ab |
| Unclassified | 12.90 ± 0.10 b | 13.55 ± 0.044 a | 13.54 ± 0.16 a | 13.08 ± 0.33 ab |
| None | 0.198 ± 0.002 a | 0.205 ± 0.005 a | 0.193 ± 0.019 a | 0.199 ± 0.006 a |
| KEGG Pathway | Treatment a | ||||
|---|---|---|---|---|---|
| p-Value | NT | NTS | T | TS | |
| Metabolism | |||||
| Carbohydrate metabolism | 0.256 | 10.62 ± 0.08 a b | 10.29 ± 0.21 a | 10.79 ± 0.48 a | 10.56 ± 0.17 a |
| Energy metabolism | 0.342 | 5.60 ± 0.05 a | 5.46 ± 0.18 a | 5.67 ± 0.19 a | 5.62 ± 0.07 a |
| Lipid metabolism | 0.030 | 4.17 ± 0.01 a | 3.89 ± 0.15 bc | 3.84 ± 0.12 c | 4.09 ± 0.14 ab |
| Nucleotide metabolism | 0.136 | 3.21 ± 0.02 a | 3.21 ± 0.04 a | 3.45 ± 0.23 a | 3.24 ± 0.12 a |
| Amino acid metabolism | 0.027 | 11.18 ± 0.12 a | 10.59 ± 0.36 b | 10.59 ± 0.13 b | 11.09 ± 0.25 a |
| Metabolism of other amino acids | 0.071 | 2.04 ± 0.03 a | 1.92 ± 0.09 a | 1.91 ± 0.02 a | 2.03 ± 0.08 a |
| Glycan biosynthesis and metabolism | 0.079 | 1.77 ± 0.03 a | 1.86 ± 0.09 a | 2.19 ± 0.33 a | 1.80 ± 0.15 a |
| Metabolism of cofactors and vitamins | 0.398 | 4.20 ± 0.03 a | 4.15 ± 0.05 a | 4.26 ± 0.13 a | 4.20 ± 0.02 a |
| Metabolism of terpenoids and polyketides | 0.084 | 2.45 ± 0.04 a | 2.26 ± 0.16 a | 2.25 ± 0.02 a | 2.41 ± 0.11 a |
| Biosynthesis of other secondary metabolites | 0.118 | 1.03 ± 0.01 a | 0.92 ± 0.07 a | 1.00 ± 0.08 a | 1.01 ± 0.02 a |
| Xenobiotics biodegradation and metabolism | 0.044 | 4.19 ± 0.06 a | 3.68 ± 0.35 ab | 3.52 ± 0.22 b | 4.09 ± 0.33 a |
| Genetic information processing | |||||
| Transcription | 0.317 | 2.46 ± 0.01 a | 2.50 ± 0.05 a | 2.52 ± 0.06 a | 2.46 ± 0.05 a |
| Translation | 0.478 | 4.12 ± 0.03 a | 4.09 ± 0.08 a | 4.34 ± 0.35 a | 4.16 ± 0.19 a |
| Folding, sorting and degradation | 0.170 | 2.19 ± 0.01 a | 2.25 ± 0.06 a | 2.31 ± 0.09 a | 2.20 ± 0.06 a |
| Replication and repair | 0.286 | 6.85 ± 0.03 a | 6.79 ± 0.09 a | 7.27 ± 0.54 a | 6.89 ± 0.30 a |
| Environmental information processing | |||||
| Membrane transport | 0.380 | 11.56 ± 0.07 a | 11.99 ± 0.13 a | 11.33 ± 0.86 a | 11.61 ± 0.16 a |
| Signal transduction | 0.182 | 2.03 ± 0.09 a | 2.32 ± 0.23 a | 1.94 ± 0.31 a | 2.01 ± 0.08 a |
| Signaling molecules and interaction | 0.283 | 0.181 ± 0.004 a | 0.193 ± 0.015 a | 0.198 ± 0.017 a | 0.182 ± 0.007 a |
| Unclassified | |||||
| Poorly characterized | 0.446 | 5.00 ± 0.04 a | 5.12 ± 0.13 a | 5.06 ± 0.06 a | 5.01 ± 0.10 a |
| Metabolic diseases | 0.027 | .081 ± 0.001 b | .089 ± 0.006 a | .091 ± 0.004 a | .082 ± 0.002 b |
| Metabolism | 0.431 | 2.48 ± 0.03 a | 2.49 ± 0.01 a | 2.53 ± 0.07 a | 2.53 ± 0.04 a |
| Genetic information processing | 0.101 | 2.16 ± 0.01 a | 2.29 ± 0.06 a | 2.27 ± 0.09 a | 2.20 ± 0.06 a |
| Enzyme families | 0.117 | 1.93 ± 0.02 a | 1.95 ± 0.04 a | 2.03 ± 0.07 a | 1.93 ± 0.06 a |
| Cellular processes and signaling | 0.028 | 3.28 ± 0.07 c | 3.68 ± 0.26 ab | 3.70 ± 0.07 a | 3.37 ± 0.17 bc |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Li, L.; Xie, J.; Coulter, J.A.; Zhang, R.; Luo, Z.; Cai, L.; Wang, L.; Gopalakrishnan, S. Soil Bacterial Diversity and Potential Functions Are Regulated by Long-Term Conservation Tillage and Straw Mulching. Microorganisms 2020, 8, 836. https://doi.org/10.3390/microorganisms8060836
Liu C, Li L, Xie J, Coulter JA, Zhang R, Luo Z, Cai L, Wang L, Gopalakrishnan S. Soil Bacterial Diversity and Potential Functions Are Regulated by Long-Term Conservation Tillage and Straw Mulching. Microorganisms. 2020; 8(6):836. https://doi.org/10.3390/microorganisms8060836
Chicago/Turabian StyleLiu, Chang, Lingling Li, Junhong Xie, Jeffrey A. Coulter, Renzhi Zhang, Zhuzhu Luo, Liqun Cai, Linlin Wang, and Subramaniam Gopalakrishnan. 2020. "Soil Bacterial Diversity and Potential Functions Are Regulated by Long-Term Conservation Tillage and Straw Mulching" Microorganisms 8, no. 6: 836. https://doi.org/10.3390/microorganisms8060836