A Differential Metabarcoding Approach to Describe Taxonomy Profiles of Bacteria and Archaea in the Saltern of Margherita di Savoia (Italy)

 ,
,  , , and
, , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. CARD-FISH Analyses
2.3. DNA Extraction
2.4. Amplicon Library Preparation and Illumina-Based Sequencing
2.5. Bioinformatic Analysis
3. Results and Discussion
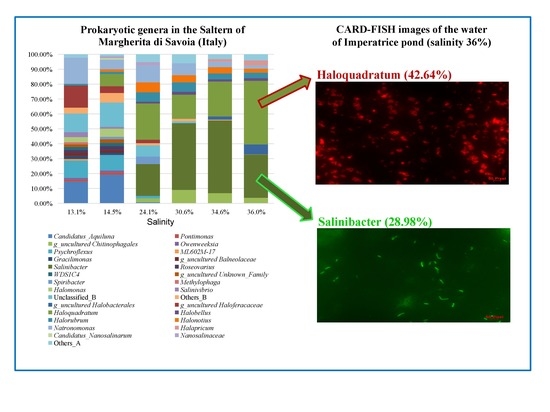
3.1. DAPI and CARD-FISH Analyses
3.2. Preparation and Sequencing of 16S rRNA Gene Metabarcoding Libraries
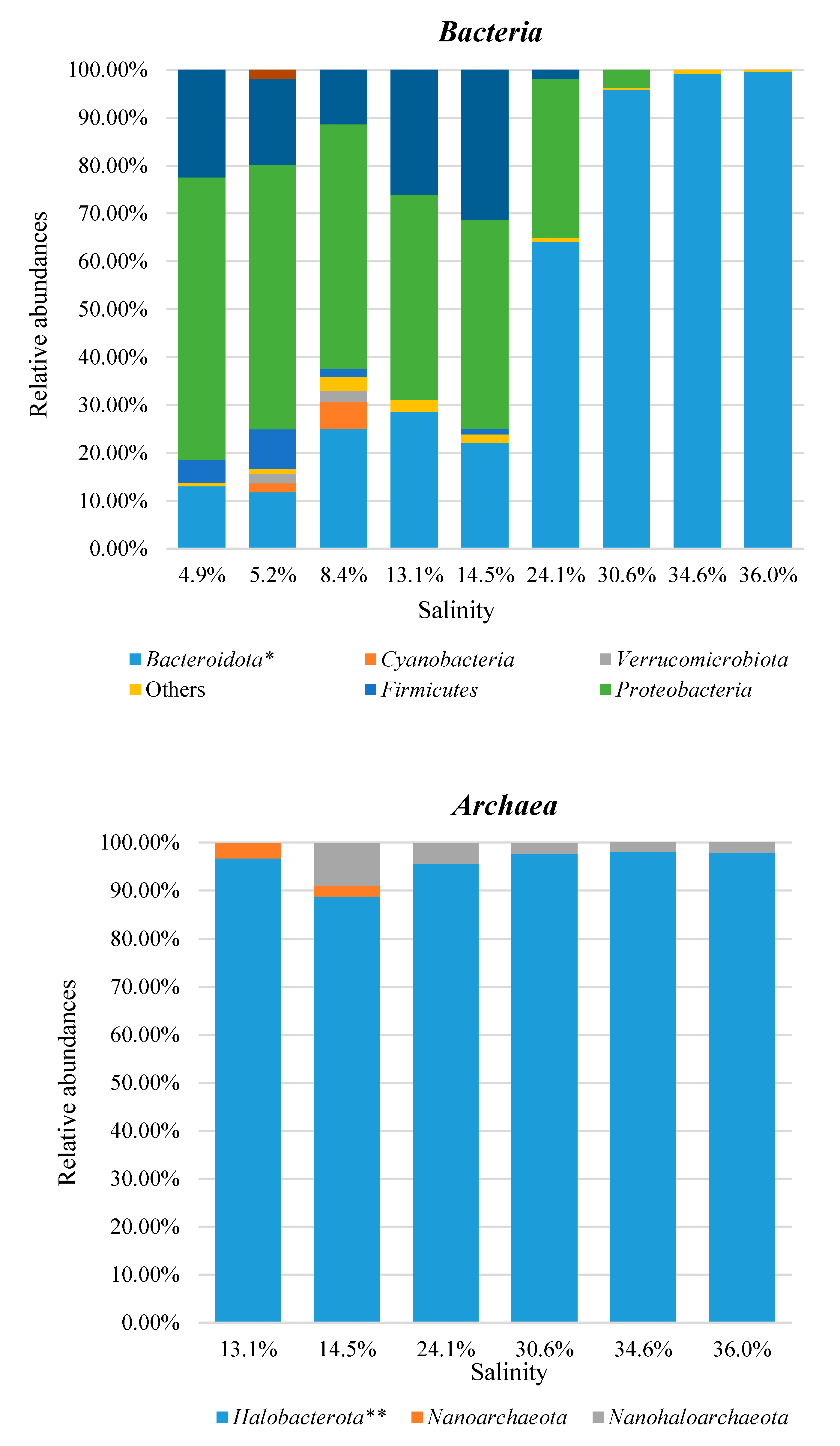
3.3. Taxonomic Profiles
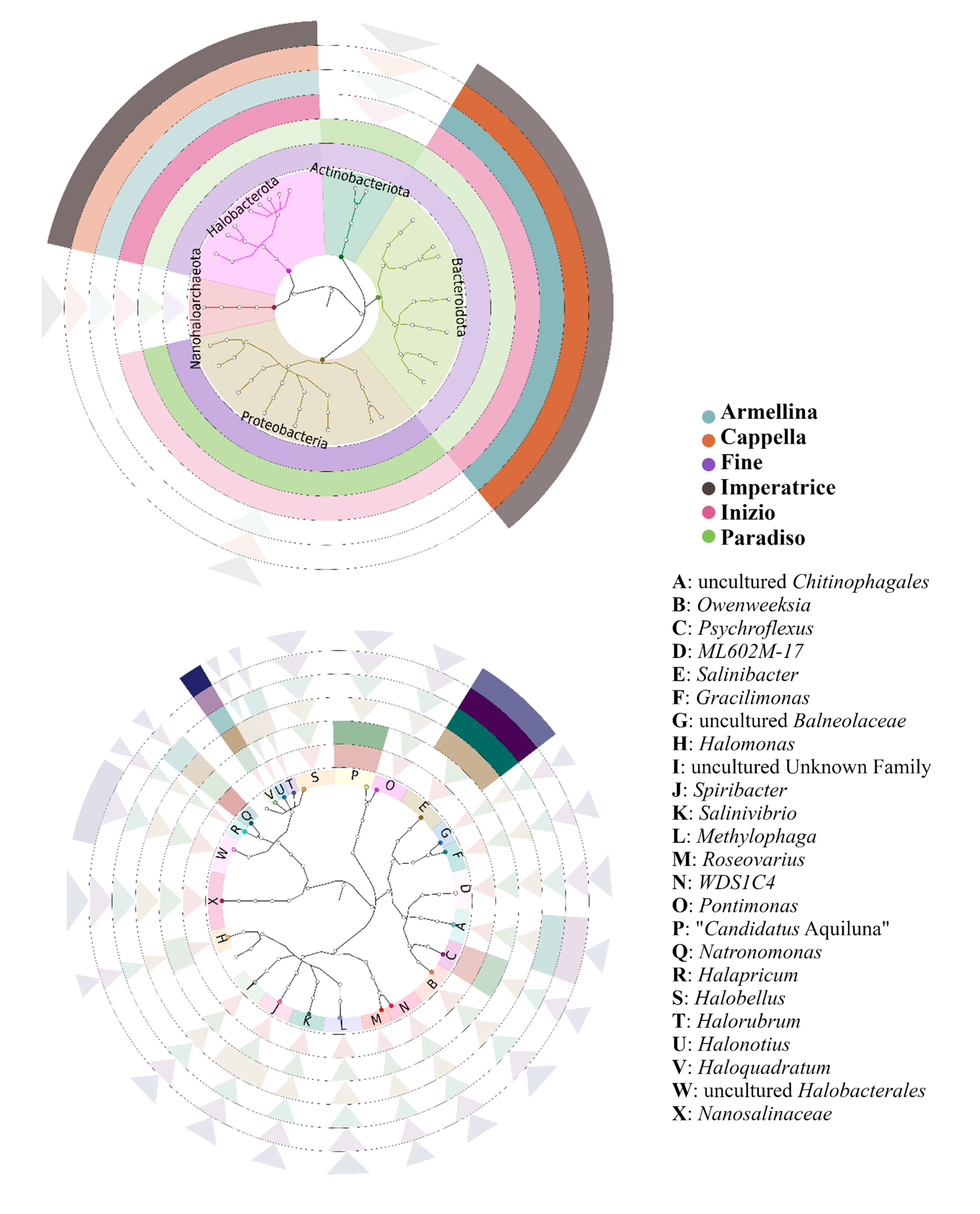
3.4. Combined Prokaryotic Populations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ollivier, B.; Caumette, P.; Garcia, J.L.; Mah, R.A. Anaerobic Bacteria from hypersaline environments. Microbiol. Rev. 1994, 58, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Ludwig, W.; Euzéby, J.; Amann, R.; Schleifer, K.-H.; Glöckner, F.O.; Rosselló-Móra, R. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Syst. Appl. Microbiol. 2010, 33, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventosa, A.; Márquez, M.C.; Ruiz-Berraquero, F.; Kocur, M. Salinicoccus roseus gen. nov., sp. nov., a New Moderately Halophilic Gram-Positive Coccus. Syst. Appl. Microbiol. 1990, 13, 29–33. [Google Scholar] [CrossRef]
- Ventosa, A.; Márquez, M.C.; Garabito, M.J.; Arahal, D.R. Moderately halophilic gram-positive bacterial diversity in hypersaline environments. Extremophiles 1998, 2, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Kamekura, M. Diversity of extremely halophilic Bacteria. Extremophiles 1998, 2, 289–295. [Google Scholar] [CrossRef]
- Oren, A. Diversity of halophilic microorganisms: Environments, phylogeny, physiology, and applications. J. Ind. Microbiol. Biotechnol. 2002, 28, 56–63. [Google Scholar] [CrossRef]
- Oren, A. Taxonomy of halophilic Archaea: Current status and future challenges. Extremophiles 2014, 18, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Pinevich, A.V.; Andronov, E.E.; Pershina, E.V.; Pinevich, A.A.; Dmitrieva, H.Y. Testing culture purity in prokaryotes: Criteria and challenges. Antonie Van Leeuwenhoek 2018, 111, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- James, K.; Cockell, S.J.; Zenkin, N. Deep sequencing approaches for the analysis of prokaryotic transcriptional boundaries and dynamics. Methods 2017, 120, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Volpicella, M.; Leoni, C.; Costanza, A.; Fanizza, I.; Placido, A.; Ceci, L.R. Genome walking by next generation sequencing approaches. Biology (Basel) 2012, 1, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.G.; Janssen, P.H.; Itoh, T.; Kamekura, M.; Li, Z.; Jensen, G.; Rodríguez-Valera, F.; Bolhuis, H.; Dyall-Smith, M.L. Haloquadratum walsbyi gen. nov., sp. nov., the square haloarchaeon of Walsby, isolated from saltern crystallizers in Australia and Spain. Int. J. Syst. Evol. Microbiol 2007, 57, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Pašić, L.; Ulrih, N.P.; Črnigoj, M.; Grabnar, M.; Velikonja, B.H. Haloarchaeal communities in the crystallizers of two adriatic solar salterns. Can. J. Microbiol. 2007, 53, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Bidle, K.; Amadio, W.; Oliveira, P.; Paulish, T.; Hicks, S.; Earnest, C. A Phylogenetic Analysis of Haloarchaea Found in a Solar Saltern. Bios 2005, 76, 89–96. [Google Scholar] [CrossRef]
- Kambourova, M.; Tomova, I.; Boyadzhieva, I.; Radchenkova, N.; Vasileva-Tonkova, E. Unusually High Archaeal Diversity in a Crystallizer Pond, Pomorie Salterns, Bulgaria, Revealed by Phylogenetic Analysis. Archaea 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anton, J.; Rossello-Mora, R.; Rodriguez-Valera, F.; Amann, R. Extremely Halophilic Bacteria in Crystallizer Ponds from Solar Salterns. Appl. Environ. Microbiol. 2000, 66, 3052–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Mora-Ruiz, M.R.; Cifuentes, A.; Font-Verdera, F.; Pérez-Fernández, C.; Farias, M.E.; González, B.; Orfila, A.; Rosselló-Móra, R. Biogeographical patterns of bacterial and archaeal communities from distant hypersaline environments. Syst. Appl. Microbiol. 2018, 41, 139–150. [Google Scholar] [CrossRef]
- Margesin, R.; Schinner, F. Potential of halotolerant and halophilic microorganisms for biotechnology. Extremophiles 2001, 5, 73–83. [Google Scholar] [CrossRef]
- DasSarma, P.; Coker, J.A.; Huse, V.; DasSarma, S. Halophiles, Industrial Applications. In Encyclopedia of Industrial Biotechnology: Bioprocess, Bioseparation, and Cell Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; ISBN 978-0-471-79930-6. [Google Scholar]
- Litchfield, C.D. Potential for industrial products from the halophilic Archaea. J. Ind. Microbiol. Biotechnol. 2011, 38, 1635–1647. [Google Scholar] [CrossRef]
- Mirete, S.; Morgante, V.; González-Pastor, J.E. Functional metagenomics of extreme environments. Curr. Opin. Biotechnol. 2016, 38, 143–149. [Google Scholar] [CrossRef]
- Kimbrel, J.A.; Ballor, N.; Wu, Y.-W.; David, M.M.; Hazen, T.C.; Simmons, B.A.; Singer, S.W.; Jansson, J.K. Microbial Community Structure and Functional Potential Along a Hypersaline Gradient. Front. Microbiol. 2018, 9, 1492. [Google Scholar] [CrossRef]
- Pernthaler, J.; Pernthaler, A.; Amann, R. Automated Enumeration of Groups of Marine Picoplankton after Fluorescence In Situ Hybridization. Appl. Environ. Microbiol. 2003, 69, 2631–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, R.I.; Krumholz, L.; Stahl, D.A. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriol. 1990, 172, 762–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daims, H.; Brühl, A.; Amann, R.; Schleifer, K.-H.; Wagner, M. The Domain-specific Probe EUB338 is Insufficient for the Detection of all Bacteria: Development and Evaluation of a more Comprehensive Probe Set. Syst. Appl. Microbiol. 1999, 22, 434–444. [Google Scholar] [CrossRef]
- Stahl, D.A.; Amann, R.I. Development and Application of Nucleic Acid Probes in Bacterial Systematics. In Stackebrandt, E. and Goodfellow, M., Eds., Nucleic Acid Techniques in Bacterial Systematics; Wiley: Chichester, NY, USA, 1991; pp. 205–242. [Google Scholar]
- Teira, E.; Reinthaler, T.; Pernthaler, A.; Pernthaler, J.; Herndl, G.J. Combining Catalyzed Reporter Deposition-Fluorescence In Situ Hybridization and Microautoradiography To Detect Substrate Utilization by Bacteria and Archaea in the Deep Ocean. Appl. Environ. Microbiol. 2004, 70, 4411–4414. [Google Scholar] [CrossRef] [Green Version]
- Manzari, C.; Chiara, M.; Costanza, A.; Leoni, C.; Volpicella, M.; Picardi, E.; D’Erchia, A.M.; Placido, A.; Trotta, M.; Horner, D.S.; et al. Draft genome sequence of Sphingobium sp. strain ba1, resistant to kanamycin and nickel ions. FEMS Microbiol. Lett. 2014, 361, 8–9. [Google Scholar] [CrossRef] [Green Version]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Stecher, B.; Chaffron, S.; Käppeli, R.; Hapfelmeier, S.; Freedrich, S.; Weber, T.C.; Kirundi, J.; Suar, M.; McCoy, K.D.; von Mering, C.; et al. Like Will to Like: Abundances of Closely Related Species Can Predict Susceptibility to Intestinal Colonization by Pathogenic and Commensal Bacteria. Plos Pathog. 2010, 6, e1000711. [Google Scholar] [CrossRef]
- Takai, K.; Horikoshi, K. Rapid Detection and Quantification of Members of the Archaeal Community by Quantitative PCR Using Fluorogenic Probes. Appl. Environ. Microbiol. 2000, 66, 5066–5072. [Google Scholar] [CrossRef] [Green Version]
- Manzari, C.; Fosso, B.; Marzano, M.; Annese, A.; Caprioli, R.; D’Erchia, A.M.; Gissi, C.; Intranuovo, M.; Picardi, E.; Santamaria, M.; et al. The influence of invasive jellyfish blooms on the aquatic microbiome in a coastal lagoon (Varano, SE Italy) detected by an Illumina-based deep sequencing strategy. Biol. Invasions 2015, 17, 923–940. [Google Scholar] [CrossRef] [Green Version]
- Leoni, C.; Ceci, O.; Manzari, C.; Fosso, B.; Volpicella, M.; Ferrari, A.; Fiorella, P.; Pesole, G.; Cicinelli, E.; Ceci, L.R. Human Endometrial Microbiota at Term of Normal Pregnancies. Genes 2019, 10, 971. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Kossaifi, J.; Gramfort, A.; Thirion, B.; Varoquaux, G. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Luan, Y.; Sun, F. Variance adjusted weighted UniFrac: A powerful beta diversity measure for comparing communities based on phylogeny. BMC Bioinform. 2011, 12, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R. R Package Version 2.3-2. Vegan: Community Ecology Package. Available online: https://cran.r-project.org/package=vegan (accessed on 19 June 2020).
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.-F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamm, J.N.; Erdmann, S.; Eloe-Fadrosh, E.A.; Angeloni, A.; Zhong, L.; Brownlee, C.; Williams, T.J.; Barton, K.; Carswell, S.; Smith, M.A.; et al. Unexpected host dependency of Antarctic Nanohaloarchaeota. Proc. Natl. Acad. Sci. USA 2019, 116, 14661–14670. [Google Scholar] [CrossRef] [Green Version]
- Oren, A. Salinibacter: An extremely halophilic bacterium with archaeal properties. FEMS Microbiol. Lett. 2013, 342, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Xia, J.; Dunlap, C.A.; Rooney, A.P.; Du, Z.-J. Psychroflexus saliphilus sp. nov., isolated from a marine solar saltern. Int. J. Syst. Evol. Microbiol. 2016, 66, 5124–5128. [Google Scholar] [CrossRef]
- Kalwasińska, A.; Deja-Sikora, E.; Burkowska-But, A.; Szabó, A.; Felföldi, T.; Kosobucki, P.; Krawiec, A.; Walczak, M. Changes in bacterial and archaeal communities during the concentration of brine at the graduation towers in Ciechocinek spa (Poland). Extremophiles 2018, 22, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.W. Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. Int. J. Syst. Evol. Microbiol 2009, 59, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Kang, I.; Lee, K.; Yang, S.-J.; Choi, A.; Kang, D.; Lee, Y.K.; Cho, J.-C. Genome Sequence of “Candidatus Aquiluna” sp. Strain IMCC13023, a Marine Member of the Actinobacteria Isolated from an Arctic Fjord. J. Bacteriol. 2012, 194, 3550–3551. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.-X.; Zhang, F.; He, J.-F.; Lee, S.H.; Qiao, Z.-Y.; Yu, Y.; Li, H.-R. Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes. Antonie Van Leeuwenhoek 2013, 103, 1309–1319. [Google Scholar] [CrossRef]
- Eronen-Rasimus, E.; Piiparinen, J.; Karkman, A.; Lyra, C.; Gerland, S.; Kaartokallio, H. Bacterial communities in Arctic first-year drift ice during the winter/spring transition: Bacterial communities in Arctic first-year drift ice. Environ. Microbiol. Rep. 2016, 8, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Gómez, B.; Díez, B.; Polz, M.F.; Arroyo, J.I.; Alfaro, F.D.; Marchandon, G.; Sanhueza, C.; Farías, L.; Trefault, N.; Marquet, P.A.; et al. Bacterial community structure in a sympagic habitat expanding with global warming: Brackish ice brine at 85–90 °N. ISME J. 2019, 13, 316–333. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, M.; Behera, P.; Kim, J.Y.; Rastogi, G. Seasonal and spatial dynamics of bacterioplankton communities in a brackish water coastal lagoon. Sci. Total Environ. 2020, 705, 134729. [Google Scholar] [CrossRef]
- Lobasso, S.; Lopalco, P.; Angelini, R.; Pollice, A.; Laera, G.; Milano, F.; Agostiano, A.; Corcelli, A. Isolation of Squarebop I bacteriorhodopsin from biomass of coastal salterns. Protein Expr. Purif. 2012, 84, 73–79. [Google Scholar] [CrossRef]
- Amoozegar, M.A.; Siroosi, M.; Atashgahi, S.; Smidt, H.; Ventosa, A. Systematics of Haloarchaea and biotechnological potential of their hydrolytic enzymes. Microbiology 2017, 163, 623–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante-Domínguez, C.; de la Haba, R.R.; Corral, P.; Sanchez-Porro, C.; Arahal, D.R.; Ventosa, A. Genome-based analyses reveal a synonymy among Halorubrum distributum Zvyagintseva and Tarasov 1989; Oren and Ventosa 1996, Halorubrum terrestre Ventosa et al. 2004, Halorubrum arcis Xu et al. 2007 and Halorubrum litoreum Cui et al. 2007. Emended description of Halorubrum distributum Zvyagintseva and Tarasov 1989; Oren and Ventosa 1996. Int. J. Syst. Evol. Microbiol. 2020, 70, 1698–1705. [Google Scholar] [CrossRef]
- Durán-Viseras, A.; Sánchez-Porro, C.; Ventosa, A. Natronomonas salsuginis sp. nov., a New Inhabitant of a Marine Solar Saltern. Microorganisms 2020, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Ventosa, A.; Fernández, A.B.; León, M.J.; Sánchez-Porro, C.; Rodriguez-Valera, F. The Santa Pola saltern as a model for studying the microbiota of hypersaline environments. Extremophiles 2014, 18, 811–824. [Google Scholar] [CrossRef]
- Ghai, R.; Pašić, L.; Fernández, A.B.; Martin-Cuadrado, A.-B.; Mizuno, C.M.; McMahon, K.D.; Papke, R.T.; Stepanauskas, R.; Rodriguez-Brito, B.; Rohwer, F.; et al. New Abundant Microbial Groups in Aquatic Hypersaline Environments. Sci. Rep. 2011, 1. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pond Name | Salinity (% w/v) | T (°C) | pH |
|---|---|---|---|
| Alma Dannata | 4.9 | 24 | 8.55 |
| Polmone | 5.2 | 26 | 8.62 |
| Zero | 8.4 | 27 | 8.07 |
| Fine | 13.1 | 25 | 7.77 |
| Paradiso | 14.5 | 27 | 7.73 |
| Inizio | 24.1 | 29 | 7.59 |
| Armellina | 30.6 | 30 | 7.46 |
| Cappella | 34.6 | 30 | 7.27 |
| Imperatrice | 36.0 | 30 | 7.20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leoni, C.; Volpicella, M.; Fosso, B.; Manzari, C.; Piancone, E.; Dileo, M.C.G.; Arcadi, E.; Yakimov, M.; Pesole, G.; Ceci, L.R. A Differential Metabarcoding Approach to Describe Taxonomy Profiles of Bacteria and Archaea in the Saltern of Margherita di Savoia (Italy). Microorganisms 2020, 8, 936. https://doi.org/10.3390/microorganisms8060936
Leoni C, Volpicella M, Fosso B, Manzari C, Piancone E, Dileo MCG, Arcadi E, Yakimov M, Pesole G, Ceci LR. A Differential Metabarcoding Approach to Describe Taxonomy Profiles of Bacteria and Archaea in the Saltern of Margherita di Savoia (Italy). Microorganisms. 2020; 8(6):936. https://doi.org/10.3390/microorganisms8060936
Chicago/Turabian StyleLeoni, Claudia, Mariateresa Volpicella, Bruno Fosso, Caterina Manzari, Elisabetta Piancone, Maria C.G. Dileo, Erika Arcadi, Michail Yakimov, Graziano Pesole, and Luigi R. Ceci. 2020. "A Differential Metabarcoding Approach to Describe Taxonomy Profiles of Bacteria and Archaea in the Saltern of Margherita di Savoia (Italy)" Microorganisms 8, no. 6: 936. https://doi.org/10.3390/microorganisms8060936