Gut Microbial Changes in Diabetic db/db Mice and Recovery of Microbial Diversity upon Pirfenidone Treatment


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Cecum DNA Extraction
2.3. 16S rDNA Analysis by MiSeq Sequencing
2.4. Processing, Filtering, and Analysis of Sequence Reads
2.5. Urine Metabolomics
2.6. Statistical Analyses for Gut Microbiota and Metabolites
3. Results and Discussion
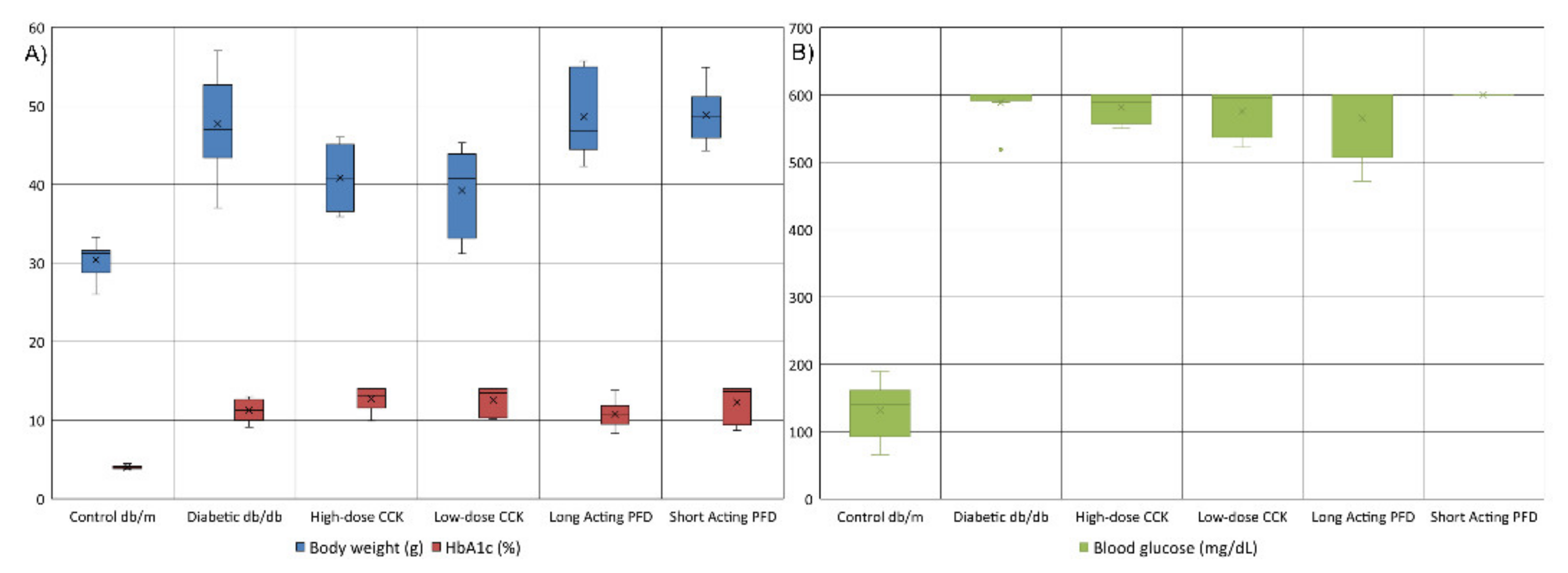
3.1. Metabolic Characteristics
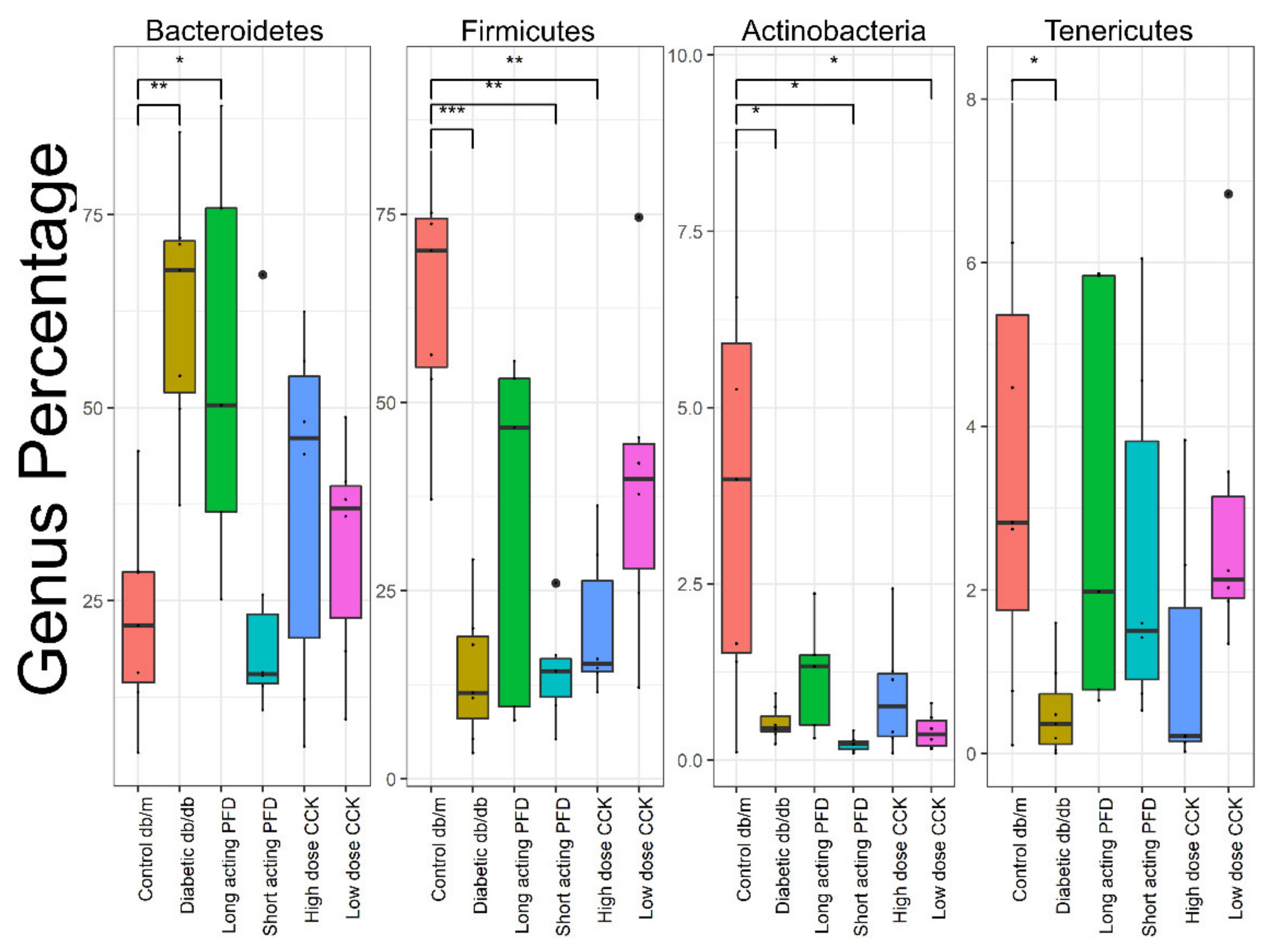
3.2. Gut Microbiota Analysis
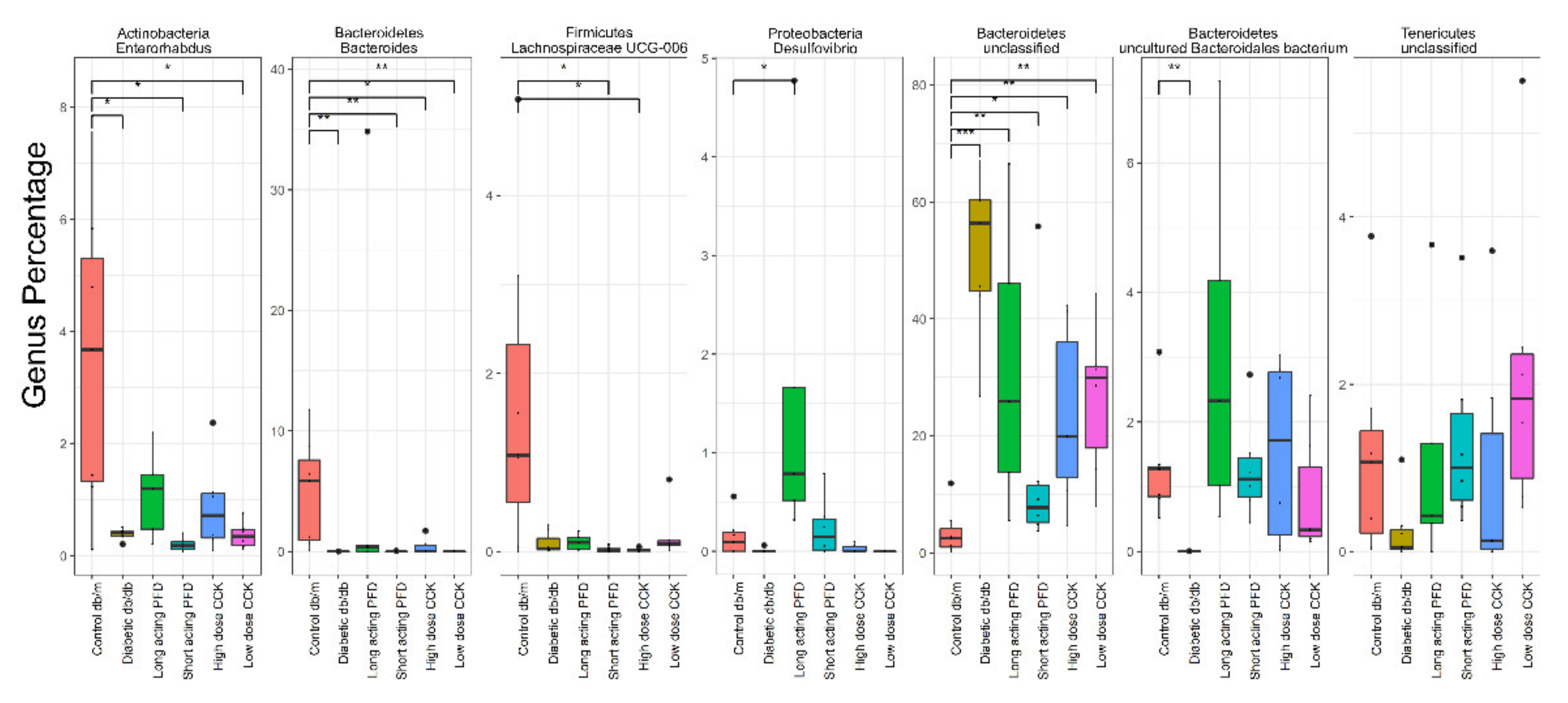
3.3. Differentially Abundant Genera
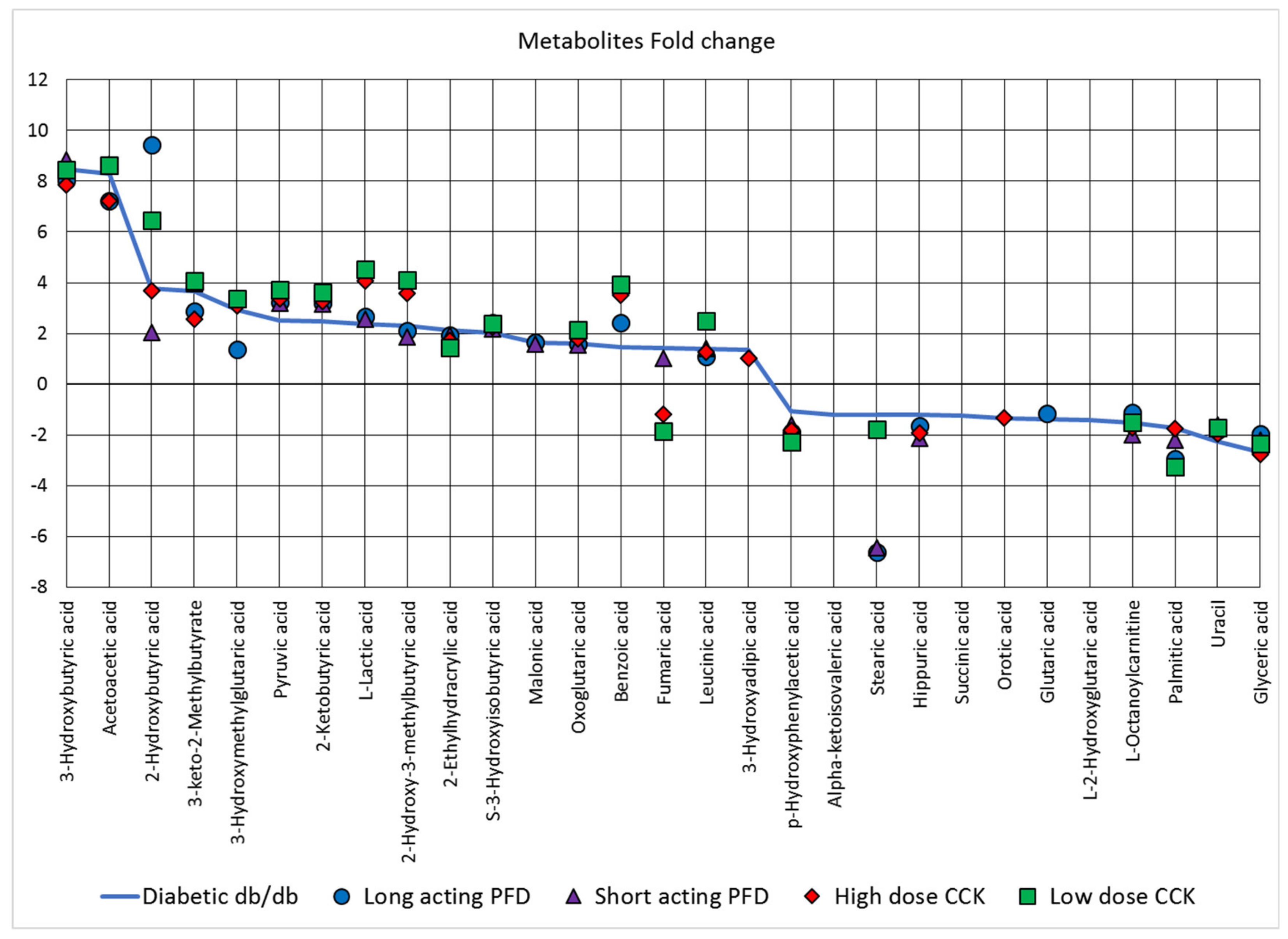
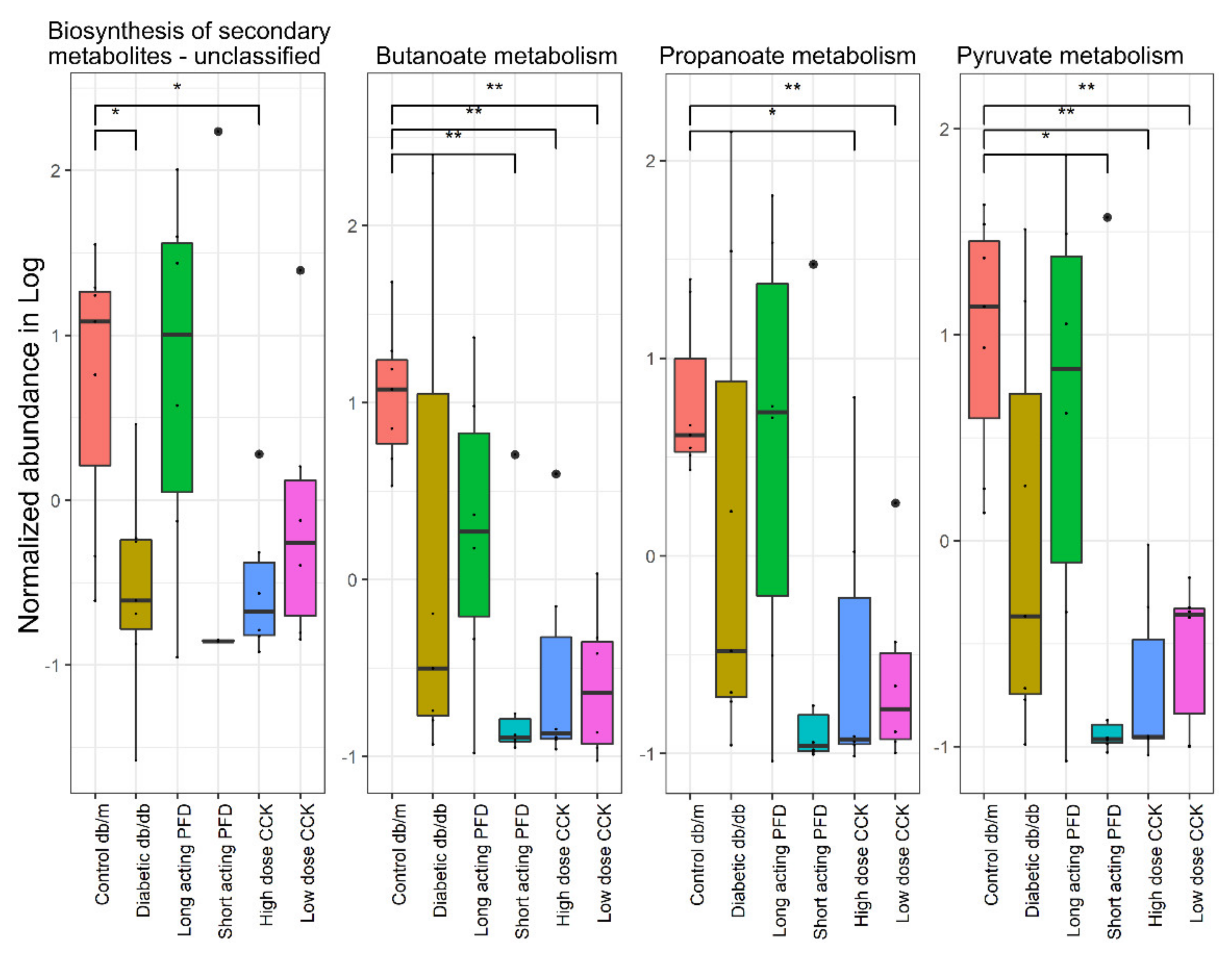
3.4. Metabolite Changes Resulting from the Drug Treatment of Diabetic db/db Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Li, R.; Bilik, D.; Brown, M.B.; Zhang, P.; Ettner, S.L.; Ackermann, R.T.; Crosson, J.C.; Herman, W.H. Medical costs associated with type 2 diabetes complications and comorbidities. Am. J. Manag. Care 2013, 19, 421–430. [Google Scholar] [PubMed]
- Slabaugh, S.L.; Curtis, B.H.; Clore, G.; Fu, H.; Schuster, D.P. Factors associated with increased healthcare costs in medicare advantage patients with type 2 diabetes enrolled in a large representative health insurance plan in the us. J. Med. Econ. 2015, 18, 106–112. [Google Scholar] [CrossRef]
- Dwyer, J.P.; Parving, H.H.; Hunsicker, L.G.; Ravid, M.; Remuzzi, G.; Lewis, J.B. Renal dysfunction in the presence of normoalbuminuria in type 2 diabetes: Results from the demand study. Cardiorenal Med. 2012, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyotylainen, T.; Hamalainen, A.M.; Peet, A.; Tillmann, V.; Poho, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Harkonen, T.; Hamalainen, A.M.; et al. Intestinal virome changes precede autoimmunity in type i diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sorensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Doumatey, A.P.; Adeyemo, A.; Zhou, J.; Lei, L.; Adebamowo, S.N.; Adebamowo, C.; Rotimi, C.N. Gut microbiome profiles are associated with type 2 diabetes in urban africans. Front. Cell Infect. Microbiol. 2020, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everard, A.; Cani, P.D. Diabetes, obesity and gut microbiota. Best Pr. Res. Clin. Gastroenterol. 2013, 27, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (tlr) activation and tlr ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetz, G.; Brown, S.M.; Hao, Y.; Tetz, V. Type 1 diabetes: An association between autoimmunity, the dynamics of gut amyloid-producing e. Coli and their phages. Sci. Rep. 2019, 9, 9685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Fisher, F.M.; Da Silva, N.F.; Khanolkar, M.; Evans, M.; Harte, A.L.; Kumar, S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E740–E747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D. Ckd impairs barrier function and alters microbial flora of the intestine: A major link to inflammation and uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2012, 21, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [Green Version]
- Tetz, G.V.; Ruggles, K.V.; Zhou, H.; Heguy, A.; Tsirigos, A.; Tetz, V. Bacteriophages as potential new mammalian pathogens. Sci. Rep. 2017, 7, 7043. [Google Scholar] [CrossRef] [Green Version]
- Mu, Q.; Kirby, J.; Reilly, C.M.; Luo, X.M. Leaky gut as a danger signal for autoimmune diseases. Front. Immunol. 2017, 8, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zhang, P.; Jiang, H.; Cheng, S. Gut bacterial translocation contributes to microinflammation in experimental uremia. Dig. Dis. Sci. 2012, 57, 2856–2862. [Google Scholar] [CrossRef]
- Ranganathan, N.; Patel, B.G.; Ranganathan, P.; Marczely, J.; Dheer, R.; Pechenyak, B.; Dunn, S.R.; Verstraete, W.; Decroos, K.; Mehta, R.; et al. In vitro and in vivo assessment of intraintestinal bacteriotherapy in chronic kidney disease. ASAIO J. 2006, 52, 70–79. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Greer, R.L.; Dong, X.; Moraes, A.C.; Zielke, R.A.; Fernandes, G.R.; Peremyslova, E.; Vasquez-Perez, S.; Schoenborn, A.A.; Gomes, E.P.; Pereira, A.C.; et al. Akkermansia muciniphila mediates negative effects of ifngamma on glucose metabolism. Nat. Commun. 2016, 7, 13329. [Google Scholar] [CrossRef]
- Belzer, C.; de Vos, W.M. Microbes inside--from diversity to function: The case of akkermansia. ISME J. 2012, 6, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 2016, 167, 1339–1353. [Google Scholar] [CrossRef] [Green Version]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; Francois, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velasquez-Mejia, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin is associated with higher relative abundance of mucin-degrading akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluznick, J.L. Gut microbiota in renal physiology: Focus on short-chain fatty acids and their receptors. Kidney Int. 2016, 90, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Oliveira, V.; Amano, M.T.; Correa-Costa, M.; Castoldi, A.; Felizardo, R.J.; de Almeida, D.C.; Bassi, E.J.; Moraes-Vieira, P.M.; Hiyane, M.I.; Rodas, A.C.; et al. Gut bacteria products prevent aki induced by ischemia-reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Furet, J.P.; Kong, L.C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Mariat, D.; Corthier, G.; Dore, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef] [Green Version]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palu, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; McCue, P.; Dunn, S.R. Diabetic kidney disease in the db/db mouse. Am. J. Physiol. Ren. Physiol. 2003, 284, F1138–F1144. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The collaborative study group. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S.; et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- RamachandraRao, S.P.; Zhu, Y.; Ravasi, T.; McGowan, T.A.; Toh, I.; Dunn, S.R.; Okada, S.; Shaw, M.A.; Sharma, K. Pirfenidone is renoprotective in diabetic kidney disease. J. Am. Soc. Nephrol. 2009, 20, 1765–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharm. Exp. 1999, 291, 367–373. [Google Scholar]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone treatment decreases transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am. J. Transplant. 2002, 2, 111–119. [Google Scholar] [CrossRef]
- Reeves, W.B.; Andreoli, T.E. Transforming growth factor beta contributes to progressive diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2000, 97, 7667–7669. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Shikata, K.; Miyasaka, K.; Okada, S.; Sasaki, M.; Kodera, R.; Hirota, D.; Kajitani, N.; Takatsuka, T.; Kataoka, H.U.; et al. Cholecystokinin plays a novel protective role in diabetic kidney through anti-inflammatory actions on macrophage: Anti-inflammatory effect of cholecystokinin. Diabetes 2012, 61, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Cottin, V.; Maher, T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2015, 24, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopala, S.V.; Yooseph, S.; Harkins, D.M.; Moncera, K.J.; Zabokrtsky, K.B.; Torralba, M.G.; Tovchigrechko, A.; Highlander, S.K.; Pieper, R.; Sender, L.; et al. Gastrointestinal microbial populations can distinguish pediatric and adolescent acute lymphoblastic leukemia (all) at the time of disease diagnosis. BMC Genom. 2016, 17, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Uparse: Highly accurate otu sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Iwai, S.; Weinmaier, T.; Schmidt, B.L.; Albertson, D.G.; Poloso, N.J.; Dabbagh, K.; DeSantis, T.Z. Piphillin: Improved prediction of metagenomic content by direct inference from human microbiomes. PLoS ONE 2016, 11, e0166104. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. Metaboanalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.H.; Jung, D.J.; Choi, E.W.; Cho, K.H.; Park, J.W.; Do, J.Y. Hba1c levels are associated with chronic kidney disease in a non-diabetic adult population: A nationwide survey (knhanes 2011–2013). PLoS ONE 2015, 10, e0145827. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are short chain fatty acids in gut microbiota defensive players for inflammation and atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Loubinoux, J.; Bronowicki, J.P.; Pereira, I.A.; Mougenel, J.L.; Faou, A.E. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol. Ecol. 2002, 40, 107–112. [Google Scholar] [CrossRef]
- Schneeberger, M.; Everard, A.; Gomez-Valades, A.G.; Matamoros, S.; Ramirez, S.; Delzenne, N.M.; Gomis, R.; Claret, M.; Cani, P.D. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci. Rep. 2015, 5, 16643. [Google Scholar] [CrossRef] [Green Version]
- Littman, D.R.; Pamer, E.G. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.C.; Derrien, M.; Isolauri, E.; de Vos, W.M.; Salminen, S. Intestinal integrity and akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl. Environ. Microbiol. 2007, 73, 7767–7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, M.; Diaz-Zepeda, C.; Eyzaguirre-Velasquez, J.; Gonzalez-Arancibia, C.; Bravo, J.A.; Julio-Pieper, M. Investigating gut permeability in animal models of disease. Front. Physiol. 2018, 9, 1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control db/m | Diabetic db/db | |||||||
|---|---|---|---|---|---|---|---|---|
| Weighted | Unweighted | Weighted | Unweighted | |||||
| PERMANOVA | R2 | p-Value | R2 | p-Value | R2 | p-Value | R2 | p-Value |
| Diabetic db/db | 0.46 | 0.001 | 0.28 | 0.001 | - | - | - | - |
| Long-acting PFD | 0.02 | 0.057 | 0.18 | 0.016 | 0.20 | 0.020 | 0.19 | 0.01 |
| Short-acting PFD | 0.38 | 0.003 | 0.21 | 0.003 | 0.43 | 0.010 | 0.18 | 0.004 |
| High-dose CCK | 0.35 | 0.003 | 0.22 | 0.005 | 0.30 | 0.003 | 0.19 | 0.003 |
| Low-dose CCK | 0.32 | 0.003 | 0.3 | 0.003 | 0.31 | 0.003 | 0.21 | 0.005 |
| ANOSI | R | p-Value | R | p-Value | R | p-Value | R | p-Value |
| Diabetic db/db | 0.856 | 0.001 | 0.502 | 0.001 | - | - | - | - |
| Long-acting PFD | 0.25 | 0.057 | 0.148 | 0.104 | 0.29 | 0.19 | 0.257 | 0.033 |
| Short-acting PFD | 0.65 | 0.003 | 0.24 | 0.014 | 0.61 | 0.007 | 0.2 | 0.026 |
| High-dose CCK | 0.68 | 0.003 | 0.266 | 0.011 | 0.414 | 0.005 | 0.312 | 0.006 |
| Low-dose CCK | 0.5 | 0.003 | 0.43 | 0.004 | 0.44 | 0.003 | 0.289 | 0.009 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, H.; Miyamoto, S.; Darshi, M.; Torralba, M.G.; Kwon, K.; Sharma, K.; Pieper, R. Gut Microbial Changes in Diabetic db/db Mice and Recovery of Microbial Diversity upon Pirfenidone Treatment. Microorganisms 2020, 8, 1347. https://doi.org/10.3390/microorganisms8091347
Singh H, Miyamoto S, Darshi M, Torralba MG, Kwon K, Sharma K, Pieper R. Gut Microbial Changes in Diabetic db/db Mice and Recovery of Microbial Diversity upon Pirfenidone Treatment. Microorganisms. 2020; 8(9):1347. https://doi.org/10.3390/microorganisms8091347
Chicago/Turabian StyleSingh, Harinder, Satoshi Miyamoto, Manjula Darshi, Manolito G. Torralba, Keehwan Kwon, Kumar Sharma, and Rembert Pieper. 2020. "Gut Microbial Changes in Diabetic db/db Mice and Recovery of Microbial Diversity upon Pirfenidone Treatment" Microorganisms 8, no. 9: 1347. https://doi.org/10.3390/microorganisms8091347