A New Method for Next-Generation Sequencing of the Full Hepatitis B Virus Genome from A Clinical Specimen: Impact for Virus Genotyping

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Virus Specimens
2.2. Ethics Approval and Consent to Participate
2.3. Sample Processing
2.4. DNA Extraction and qPCR
2.5. Primer Design and Full-Genome PCR Reactions
2.6. Next-Generation Sequencing
2.7. NGS Data Processing, Mapping, and Genome Comparisons
2.8. Phylogenetic Analysis
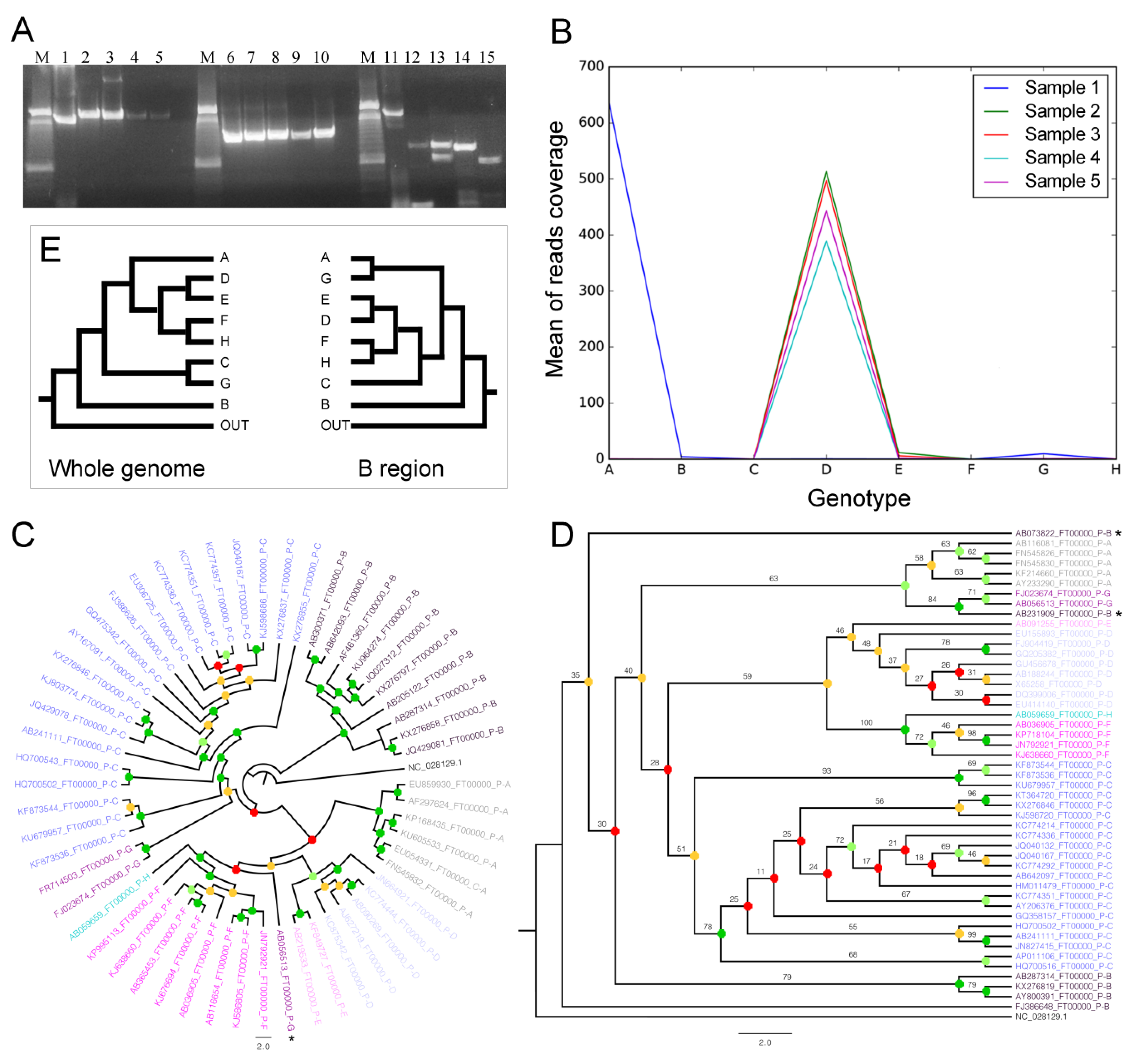
3. Results
3.1. DNA Quantification and qPCR
3.2. Full-Genome Amplification by NestedPCR
3.3. Library Quantification
3.4. NGS Data Analyses and Genome Coverage
3.4.1. Cleaning
3.4.2. Genomic Mapping, Comparisons, and Genotyping
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 4 September 2018).
- Ganem, D.; Varmus, H.E. The molecular biology of the hepatitis B viruses. Annu. Rev. Biochem. 1987, 56, 651–693. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res. 2016, 2, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; Couroucé, A.M.; Coursaget, P.; Echevarria, J.M.; Lee, S.D.; Mushahwar, I.K.; Robertson, B.H.; Locarnini, S.; Magnius, L.O. Genetic diversity of hepatitis B virus strains derived worldwide: Genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004, 47, 289–309. [Google Scholar] [CrossRef]
- Okamoto, H.; Tsuda, F.; Sakugawa, H.; Sastrosoewignjo, R.I.; Imai, M.; Miyakawa, Y.; Mayumi, M. Typing Hepatitis B Virus by Homology in Nucleotide Sequence: Comparison of Surface Antigen Subtypes. J. Gen. Virol. 1988, 69, 2575–2583. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Wu, C.C.; Chen, X.W.; Li, X.; Li, J.; Lu, M.J. Genetic variation of hepatitis B virus and its significance for pathogenesis. World J. Gastroenterol. 2016, 22, 126–144. [Google Scholar] [CrossRef]
- Huy, T.T.T.; Ngoc, T.T.; Abe, K. New Complex Recombinant Genotype of Hepatitis B Virus Identified in Vietnam. J. Virol. 2008, 82, 5657–5663. [Google Scholar] [CrossRef] [Green Version]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef] [Green Version]
- Sertoz, R.Y.; Erensoy, S.; Pas, S.; Akarca, U.S.; Ozgenc, F.; Yamazhan, T.; Ozacar, T.; Niesters, H.G.M. Comparison of Sequence Analysis and INNO-LiPA HBV DR Line Probe Assay in Patients with Chronic Hepatitis B. J. Chemother. 2005, 17, 514–520. [Google Scholar] [CrossRef] [Green Version]
- Solmone, M.; Vincenti, D.; Prosperi, M.C.F.; Bruselles, A.; Ippolito, G.; Capobianchi, M.R. Use of massively parallel ultradeep pyrosequencing to characterize the genetic diversity of hepatitis B virus in drug-resistant and drug-naive patients and to detect minor variants in reverse transcriptase and hepatitis B S antigen. J. Virol. 2009, 83, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Lampe, E.; Mello, F.C.A.; do Espírito-Santo, M.P.; Oliveira, C.M.C.; Bertolini, D.A.; Gonçales, N.S.L.; Moreira, R.C.; Fernandes, C.A.S.; Nascimento, H.C.L.; Grotto, R.M.T.; et al. Nationwide overview of the distribution of hepatitis B virus genotypes in Brazil: A 1000-sample multicentre study. J. Gen. Virol. 2017, 98, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Quiñones-Mateu, M.E.; Avila, S.; Reyes-Teran, G.; Martinez, M.A. Deep sequencing: Becoming a critical tool in clinical virology. J. Clin. Virol. 2014, 61, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Caballero, A.; Gregori, J.; Homs, M.; Tabernero, D.; Gonzalez, C.; Quer, J.; Blasi, M.; Casillas, R.; Nieto, L.; Riveiro-Barciela, M.; et al. Complex genotype mixtures analyzed by deep sequencing in two different regions of Hepatitis B virus. PLoS ONE 2015, 10, e0144816. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, Y.; Mei, Y.; Wang, Y.; Liu, T.; Guan, Y.; Tan, D.; Liang, Y.; Yang, L.; Yi, X. Analysis of hepatitis B virus genotyping and drug resistance gene mutations based on massively parallel sequencing. J. Virol. Methods 2013, 193, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.R.; Sede, M.; Manrique, J.M.; Quarleri, J. Hepatitis B virus resistance substitutions: Long-term analysis by next-generation sequencing. Arch. Virol. 2016, 161, 2885–2891. [Google Scholar] [CrossRef] [PubMed]
- Lowe, C.F.; Merrick, L.; Harrigan, P.R.; Mazzulli, T.; Sherlock, C.H.; Ritchie, G. Implementation of next-generation sequencing for hepatitis B resistance and genotyping in a clinical microbiology laboratory. J. Clin. Microbiol. 2015, 54, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.C.; Lin, C.P.; Cheng, C.P.; Ho, C.H.; Lan, K.L.; Cheng, J.H.; Yen, C.J.; Cheng, P.N.; Wu, I.C.; Li, I.C.; et al. Aligning to the sample-specific reference sequence to optimize the accuracy of next-generation sequencing analysis for hepatitis B virus. Hepatol. Int. 2016, 10, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.P.d.T.; Levi, J.E.; Lemos, M.F.; Calux, S.J.; Oba, I.T.; Moreira, R.C. An in-house real-time polymerase chain reaction: Standardisation and comparison with the cobas amplicor HBV monitor and cobas ampliprep/cobas taqman HBV tests for the quantification of hepatitis B virus DNA. Mem. Inst. Oswaldo Cruz 2016, 111, 134–140. [Google Scholar] [CrossRef] [Green Version]
- The Hepatitis B Virus Database. Available online: https://hbvdb.lyon.inserm.fr/HBVdb/. (accessed on 1 December 2018).
- Hayer, J.; Jadeau, F.; Deléage, G.; Kay, A.; Zoulim, F.; Combet, C. HBVdb: A knowledge database for Hepatitis B Virus. Nucleic Acids Res. 2013, 41, 566–570. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ali, M.M.; Hasan, F.; Ahmad, S.; Al-Nakib, W. Comparative evaluation of INNO-LiPA HBV assay, direct DNA sequencing and subtractive PCR-RFLP for genotyping of clinical HBV isolates. Virol. J. 2010, 7, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Osiowy, C.; Giles, E. Evaluation of the INNO-LiPA HBV Genotyping Assay for Determination of Hepatitis B Virus Genotype. J. Clin. Microbiol. 2003, 41, 5473–5477. [Google Scholar] [CrossRef] [Green Version]
- Mercier, M.; Laperche, S.; Girault, A.; Sureau, C.; Servant-Delmas, A. Overestimation of incidence of hepatitis B virus mixed-genotype infections by use of the new line probe INNO-LiPA genotyping assay. J. Clin. Microbiol. 2011, 49, 1154–1156. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Orito, E.; Gish, R.G.; Sugauchi, F.; Suzuki, S.; Ueda, R.; Miyakawa, Y.; Mizokami, M. Characteristics of Hepatitis B Virus Isolates of Genotype G and Their Phylogenetic Differences from the Other Six Genotypes (A through F). J. Virol. 2002, 76, 6131–6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Sample ID | Initial Viral Load (UI/mL and Log) a | Viral Load after DNAse (UI/mL and Log) b | dsDNA (ng/μL) c | Genotype (INNO-LiPA) d |
|---|---|---|---|---|
| 1 | 536,525/5.73 | 910,000/5.95 | ND | A |
| 2 | 1,439,366/6.16 | 1,700,000/6.23 | ND | D/G |
| 3 | 120,781/5.08 | 330,000/5.52 | ND | D/G |
| 4 | 894/2.95 | 2200/3.34 | ND | A/D |
| 5 | 2654/3.42 | 3900/3.59 | ND | D/G |
| Primers | Sequence (5′–3′) | Genome Binding Position a | Amplicon Size (bp) |
|---|---|---|---|
| AF | AAG AAC TCC CTC GCC TC | 2374–2390 | |
| AR | GAT GAT GGG ATG GGA ATA CAR GTG | 595–618 | 1460 |
| BF | GGT ATG TTG CCC GTT TGT CC | 458–477 | |
| BR | GCW AGG AGT TCC GCA GTA TGG | 1266–1286 | 829 |
| CF | GCT GAY GCA ACC CCC ACT G | 1186–1204 | |
| CR | CTG CGA GGC GAG GGA GTT C | 2376–2394 | 1208 |
| Before Cleaning | After Cleaning | ||||
|---|---|---|---|---|---|
| Sample ID | N. R. a | Sequence Range | N. R. a | Sequence Range | P.S.R. b |
| 1 | 729,656 | 35–151 | 265,092 | 16–135 | 36.33 |
| 2 | 654,788 | 35–151 | 236,472 | 16–135 | 36.11 |
| 3 | 716,362 | 35–151 | 229,617 | 16–135 | 32.05 |
| 4 | 585,970 | 35–151 | 215,458 | 16–135 | 36.76 |
| 5 | 672,119 | 35–151 | 240,780 | 16–135 | 35.82 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hebeler-Barbosa, F.; Wolf, I.R.; Valente, G.T.; Mello, F.C.d.A.; Lampe, E.; Pardini, M.I.d.M.C.; Grotto, R.M.T. A New Method for Next-Generation Sequencing of the Full Hepatitis B Virus Genome from A Clinical Specimen: Impact for Virus Genotyping. Microorganisms 2020, 8, 1391. https://doi.org/10.3390/microorganisms8091391
Hebeler-Barbosa F, Wolf IR, Valente GT, Mello FCdA, Lampe E, Pardini MIdMC, Grotto RMT. A New Method for Next-Generation Sequencing of the Full Hepatitis B Virus Genome from A Clinical Specimen: Impact for Virus Genotyping. Microorganisms. 2020; 8(9):1391. https://doi.org/10.3390/microorganisms8091391
Chicago/Turabian StyleHebeler-Barbosa, Flavia, Ivan Rodrigo Wolf, Guilherme Targino Valente, Francisco Campello do Amaral Mello, Elisabeth Lampe, Maria Inês de Moura Campos Pardini, and Rejane Maria Tommasini Grotto. 2020. "A New Method for Next-Generation Sequencing of the Full Hepatitis B Virus Genome from A Clinical Specimen: Impact for Virus Genotyping" Microorganisms 8, no. 9: 1391. https://doi.org/10.3390/microorganisms8091391