A Genome-Scale Metabolic Model of Thalassiosira pseudonana CCMP 1335 for a Systems-Level Understanding of Its Metabolism and Biotechnological Potential


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reconstruction of a Genome-Scale Metabolic Model
2.2. Draft Model
2.3. Gap-Filling of the Model
2.4. Compartments in the Model
2.5. Energy Requirements
2.6. Measurement of Biomass Composition for Setting Up the Biomass Equation
2.7. Model Simulations
2.8. Simulations of Si+ Limited Conditions
2.9. Reaction Essentiality Analysis
2.10. Maximum Theoretical Yields for Various Industrial Compounds
3. Results
3.1. Gap Filling
3.2. General Properties of the Model
3.3. Comparison of iThaps987 Model with P. tricornutum Model, iLB1025
3.4. Model Simulations
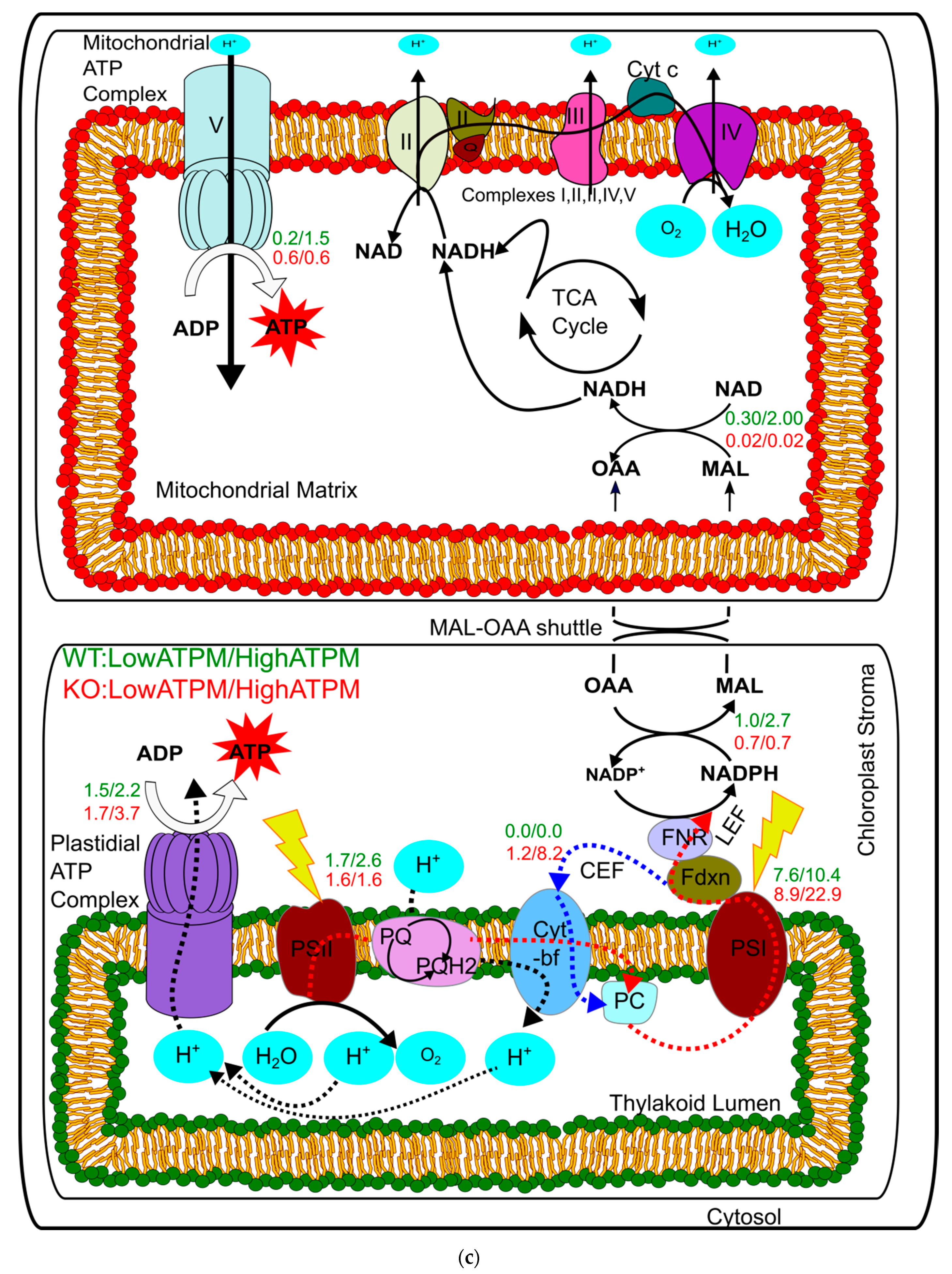
3.5. Light-Driven Generation of ATP and NADPH: Light Reaction
3.6. Carbon Fixation by the CBB Cycle (C3 Cycle): Dark Reaction
3.7. Simulation of Si+ Limited Condition
3.8. Effect of Maintenance Energy on Flux Predictions
3.9. Photoautotrophic Production of Industrially-Relevant Compounds
3.10. Reaction Deletion Analysis to Identify Essential Reactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tréguer, P.; Nelson, D.M.; Van Bennekom, A.J.; Demaster, D.J.; Leynaert, A.; Quéguiner, B. The silica balance in the world ocean: A reestimate. Science 1995, 268, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Tréguer, P.; Brzezinski, M.A.; Leynaert, A.; Quéguiner, B. Production and dissolution of biogenic silica in the ocean: Revised global estimates, comparison with regional data and relationship to biogenic sedimentation. Glob. Biogeochem. Cycles 1995, 9, 359–372. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, J.P.; Wu, C.F.; Wang, J.H. Fucoxanthin, a marine carotenoid present in brown seaweeds and diatoms: Metabolism and bioactivities relevant to human health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.; Kapoore, R.V.; Vaidyanathan, S. Phaeodactylum tricornutum: A Diatom Cell Factory. Trends Biotechnol. 2020, 38, 606–622. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Davis, A.K.; Smith, S.R.; Traller, J.C.; Abbriano, R. The place of diatoms in the biofuels industry. Biofuels 2012, 3, 221–240. [Google Scholar] [CrossRef] [Green Version]
- Chiovitti, A.; Molino, P.; Crawford, S.A.; Teng, R.; Spurck, T.; Wetherbee, R. The glucans extracted with warm water from diatoms are mainly derived from intracellular chrysolaminaran and not extracellular polysaccharides. Eur. J. Phycol. 2004, 39, 117–128. [Google Scholar] [CrossRef]
- Sayanova, O.; Mimouni, V.; Ulmann, L.; Morant-Manceau, A.; Pasquet, V.; Schoefs, B.; Napier, J.A. Modulation of lipid biosynthesis by stress in diatoms. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160407. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Marella, T.K.; Tao, L.; Li, R.; Tiwari, A.; Li, G. Optimization of growth conditions and fatty acid analysis for three freshwater diatom isolates. Phycol. Res. 2017, 65, 177–187. [Google Scholar] [CrossRef]
- Marella, T.K.; Tiwari, A. Marine diatom Thalassiosira weissflogii based biorefinery for co-production of eicosapentaenoic acid and fucoxanthin. Bioresour. Technol. 2020, 307, 123245. [Google Scholar] [CrossRef]
- Richthammer, P.; Börmel, M.; Brunner, E.; van Pée, K.-H. Biomineralization in Diatoms: The Role of Silacidins. ChemBioChem 2011, 12, 1362–1366. [Google Scholar] [CrossRef]
- Delalat, B.; Sheppard, V.C.; Rasi Ghaemi, S.; Rao, S.; Prestidge, C.A.; McPhee, G.; Rogers, M.L.; Donoghue, J.F.; Pillay, V.; Johns, T.G.; et al. Targeted drug delivery using genetically engineered diatom biosilica. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Trentacoste, E.M.; Shrestha, R.P.; Smith, S.R.; Glé, C.; Hartmann, A.C.; Hildebrand, M.; Gerwick, W.H. Metabolic engineering of lipid catabolism increases microalgal lipid accumulation without compromising growth. Proc. Natl. Acad. Sci. USA 2013, 110, 19748–19753. [Google Scholar] [CrossRef] [Green Version]
- Cook, O.; Hildebrand, M. Enhancing LC-PUFA production in Thalassiosira pseudonana by overexpressing the endogenous fatty acid elongase genes. J. Appl. Phycol. 2016, 28, 897–905. [Google Scholar] [CrossRef]
- Manandhar-Shrestha, K.; Hildebrand, M. Characterization and manipulation of a DGAT2 from the diatom Thalassiosira pseudonana: Improved TAG accumulation without detriment to growth, and implications for chloroplast TAG accumulation. Algal Res. 2015, 12, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.R.; Ahmad, A.; Srivastava, S.; Jaffar Ali, B.M. Reconstruction and analysis of a genome-scale metabolic model of Nannochloropsis gaditana. Algal Res. 2017, 26, 354–364. [Google Scholar] [CrossRef]
- Poolman, M.G.; Kundu, S.; Shaw, R.; Fell, D.A. Responses to Light Intensity in a Genome-Scale Model of Rice Metabolism. Plant. Physiol. 2013, 162, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, H.B.; Fell, D.A.; Rossell, S.; Jensen, P.R.; Woodward, M.J.; Thorndahl, L.; Jelsbak, L.; Olsen, J.E.; Raghunathan, A.; Daefler, S.; et al. Identification of potential drug targets in Salmonella enterica sv. Typhimurium using metabolic modelling and experimental validation. Microbiology 2014, 160, 1252–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, I.; Palsson, B. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [Green Version]
- Fell, D.A.; Poolman, M.G.; Gevorgyan, A. Building and analysing genome-scale metabolic models. Biochem. Soc. Trans. 2010, 38, 1197–1201. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Hartman, H.B.; Krishnakumar, S.; Fell, D.A.; Poolman, M.G.; Srivastava, S. A Genome Scale Model of Geobacillus thermoglucosidasius (C56-YS93) reveals its biotechnological potential on rice straw hydrolysate. J. Biotechnol. 2017, 251, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Monk, J.M.; Koza, A.; Campodonico, M.A.; Machado, D.; Seoane, J.M.; Palsson, B.O.; Herrgård, M.J.; Feist, A.M. Multi-omics Quantification of Species Variation of Escherichia coli Links Molecular Features with Strain Phenotypes. Cell Syst. 2016, 3, 238–251. e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safak Yilmaz, L.; Walhout, A.J.M. A Caenorhabditis elegans Genome-Scale Metabolic Network Model. Cell Syst. 2016, 2, 297–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Gomes de Oliveira Dal’Molin, C.; Quek, L.-E.; Palfreyman, R.W.; Nielsen, L.K. AlgaGEM—A genome-scale metabolic reconstruction of algae based on the Chlamydomonas reinhardtii genome. BMC Genom. 2011, 12, S5. [Google Scholar]
- Imam, S.; Schäuble, S.; Valenzuela, J.; Lõpez García De Lomana, A.; Carter, W.; Price, N.D.; Baliga, N.S. A refined genome-scale reconstruction of Chlamydomonas metabolism provides a platform for systems-level analyses. Plant. J. 2015, 84, 1239–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuñiga, C.; Li, C.-T.; Huelsman, T.; Levering, J.; Zielinski, D.C.; McConnell, B.O.; Long, C.P.; Knoshaug, E.P.; Guarnieri, M.T.; Antoniewicz, M.R.; et al. Genome-Scale Metabolic Model for the Green Alga Chlorella vulgaris UTEX 395 Accurately Predicts Phenotypes under Autotrophic, Heterotrophic, and Mixotrophic Growth Conditions. Plant. Physiol. 2016, 172, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Loira, N.; Mendoza, S.; Paz Cortés, M.; Rojas, N.; Travisany, D.; Di Genova, A.; Gajardo, N.; Ehrenfeld, N.; Maass, A. Reconstruction of the microalga Nannochloropsis salina genome-scale metabolic model with applications to lipid production. BMC Syst. Biol. 2017, 11, 66. [Google Scholar] [CrossRef]
- Levering, J.; Broddrick, J.; Dupont, C.L.; Peers, G.; Beeri, K.; Mayers, J.; Gallina, A.A.; Allen, A.E.; Palsson, B.O.; Zengler, K. Genome-Scale Model Reveals Metabolic Basis of Biomass Partitioning in a Model Diatom. PLoS ONE 2016, 11, e0155038. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Fabris, M.; Baart, G.; Kim, M.K.; Goossens, A.; Vyverman, W.; Falkowski, P.G.; Lun, D.S. Flux balance analysis of primary metabolism in the diatom Phaeodactylum tricornutum. Plant. J. 2016, 85, 161–176. [Google Scholar] [CrossRef] [Green Version]
- LEWIN, J.C. The taxonomic position of Phaeodactylum tricornutum. J. Gen. Microbiol. 1958, 18, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Kociolek, J.P.; Theriot, E.C.; Williams, D.M.; Julius, M.; Stoermer, E.F.; Kingston, J.C. Centric and araphid diatoms. In Freshwater Algae of North America: Ecology and Classification; Elsevier Inc.: Amsterdam, The Netherlands, 2015; pp. 653–708. ISBN 9780123858771. [Google Scholar]
- Bowler, C.; Falciatore, A. Phaeodactylum tricornutum. Trends Genet. 2019, 35, 706–707. [Google Scholar] [CrossRef]
- Brembu, T.; Mühlroth, A.; Alipanah, L.; Bones, A.M. The effects of phosphorus limitation on carbon metabolism in diatoms. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160406. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Gu, W.; Wu, S.; Huang, A.; He, L.; Xie, X.; Gao, S.; Zhang, B.; Niu, J.; Peng Lin, A.; et al. Silicon enhances the growth of Phaeodactylum tricornutum Bohlin under green light and low temperature. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, A.D.; Meichenin, A.; Shi, J.; Pan, K.; Bowler, C. Genetic and phenotypic characterization of Phaeodactylum tricornutum (Bacillariophyceae) accessions 1. J. Phycol. 2007, 43, 992–1009. [Google Scholar] [CrossRef]
- Poolman, M.G.; Miguet, L.; Sweetlove, L.J.; Fell, D.A. A Genome-Scale Metabolic Model of Arabidopsis and Some of Its Properties. Plant. Physiol. 2009, 151, 1570–1581. [Google Scholar] [CrossRef] [Green Version]
- Poolman, M.G. ScrumPy: Metabolic modelling with Python. Syst. Biol. 2006, 153, 375–378. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. In Briefings in Bioinformatics; Oxford University Press: Oxford, UK, 2019; Volume 20, pp. 1085–1093. [Google Scholar]
- MetaCyc Database of Metabolic Pathways and Enzymes|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/46/D1/D633/4559117 (accessed on 21 May 2020).
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Pathania, R.; Srivastava, S. Biochemical characteristics and a genome-scale metabolic model of an indian euryhaline cyanobacterium with high polyglucan content. Metabolites 2020, 10, 177. [Google Scholar] [CrossRef]
- King, B.R.; Vural, S.; Pandey, S.; Barteau, A.; Guda, C. NgLOC: Software and web server for predicting protein subcellular localization in prokaryotes and eukaryotes. BMC Res. Notes 2012, 5, 351. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.-R.; Lakshmanan, M.; Aggarwal, S.; Song, J.-W.; Karimi, I.A.; Lee, D.-Y.; Park, J.-B. Genome-scale metabolic network reconstruction and in silico flux analysis of the thermophilic bacterium Thermus thermophilus HB27. Microb. Cell Fact. 2014, 13, 61. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Cheung, C.Y.M.; Poolman, M.G.; Hilbers, P.A.J.; Van Riel, N.A.W. A genome-scale metabolic network reconstruction of tomato (Solanum lycopersicum L.) and its application to photorespiratory metabolism. Plant. J. 2016, 85, 289–304. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Xu, M.; Di, X.; Brynjolfsson, S.; Fu, W. Exploring valuable lipids in diatoms. Front. Mar. Sci. 2017, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Athanasakoglou, A.; Kampranis, S.C. Diatom isoprenoids: Advances and biotechnological potential. Biotechnol. Adv. 2019, 37, 107417. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Manandhar-Shrestha, K.; Abbriano, R. Effects of chrysolaminarin synthase knockdown in the diatom Thalassiosira pseudonana: Implications of reduced carbohydrate storage relative to green algae. Algal Res. 2017, 23, 66–77. [Google Scholar] [CrossRef]
- Klanchui, A.; Dulsawat, S.; Chaloemngam, K.; Cheevadhanarak, S.; Prommeenate, P.; Meechai, A. An improved genome-scale metabolic model of Arthrospira platensis C1 (iAK888) and its application in glycogen overproduction. Metabolites 2018, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Direct Impact of Silicate on the Photosynthetic Performance of the Diatom Thalassiosira weissflogii Assessed by On- and Off-Line PAM Fluorescence Measurements|Journal of Plankton Research|Oxford Academic. Available online: https://academic.oup.com/plankt/article/21/2/269/1432210 (accessed on 24 July 2020).
- Bailleul, B.; Berne, N.; Murik, O.; Petroutsos, D.; Prihoda, J.; Tanaka, A.; Villanova, V.; Bligny, R.; Flori, S.; Falconet, D.; et al. Energetic coupling between plastids and mitochondria drives CO 2 assimilation in diatoms. Nature 2015, 524, 366–369. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.; Granum, E.; Leegood, R.C.; Raven, J.A. C3 and C4 pathways of photosynthetic carbon assimilation in marine diatoms are under genetic, not environmental, control. Plant. Physiol. 2007, 145, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.E.; Dupont, C.L.; Oborník, M.; Horák, A.; Nunes-Nesi, A.; McCrow, J.P.; Zheng, H.; Johnson, D.A.; Hu, H.; Fernie, A.R.; et al. Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature 2011, 473, 203–207. [Google Scholar] [CrossRef]
- Dyhrman, S.T.; Jenkins, B.D.; Rynearson, T.A.; Saito, M.A.; Mercier, M.L.; Alexander, H.; Whitney, L.P.; Drzewianowski, A.; Bulygin, V.V.; Bertrand, E.M.; et al. The Transcriptome and Proteome of the Diatom Thalassiosira pseudonana Reveal a Diverse Phosphorus Stress Response. PLoS ONE 2012, 7, e33768. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, R.N.; Bopp, S.K.; Lettieri, T. Transcriptomics Responses in Marine Diatom Thalassiosira pseudonana Exposed to the Polycyclic Aromatic Hydrocarbon Benzo[a]pyrene. PLoS ONE 2011, 6, e26985. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, R.P.; Tesson, B.; Norden-Krichmar, T.; Federowicz, S.; Hildebrand, M.; Allen, A.E. Whole transcriptome analysis of the silicon response of the diatom Thalassiosira pseudonana. BMC Genom. 2012, 13, 499. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytosol | Chloroplast | Mitochondrion | Transport | |
|---|---|---|---|---|
| Reactions | 1125 | 887 | 377 | 88 |
| Metabolites | 1024 | 984 | 467 | - |
| Microorganism | P. tricornutum | P. tricornutum | T. pseudonana |
|---|---|---|---|
| Genome size (Mbp) | 27.45 | 27.45 | 32.61 |
| Genes in the model | 1027 (10,392) | - | 987 |
| Number of reactions/metabolites | 2156 */2172 | 849/587 | 2477/2456 |
| Compartments | 6 | 4 | 3 |
| Reference | [28] | [29] | This study |
| S.N | Product | Yield Photoautotrophic (mol/mol carbon) | No. of Added Reactions | Added Genes |
|---|---|---|---|---|
| Native | iThaps987 | |||
| 1 | Malate | 0.22 | 0 | − |
| 2 | Succinate | 0.22 | 0 | − |
| 3 | Citrate | 0.14 | 0 | − |
| Non-Native | ||||
| 4 | 2-MethylSuccinate | 0.17 | 4 | cimA, leuC, leuD, ER |
| 5 | Styrene | 0.11 | 2 | (PAL, PAL1, encP), FDC1 |
| 6 | iso-butanol | 0.22 | 1 | kivD |
| 7 | Valencene | 0.06 | 2 | Valcs, Vals, Tps1 |
| 8 | Farnesyl-PP | 0.06 | afs | |
| 9 | Naringenin | 0.06 | 3 | PAL, (CHI1 and CHI2),CHS-A |
| 10 | 5-Amino-Levulinate | 0.17 | 1 | hemA or HemT |
| 11 | Isoprene | 0.17 | 4 | Isps, IspG, IspH * (* catalyses 2 reactions) |
| 12 | PHB | 0.22 | 2 | phbB, phbC |
| 14 | Citramalate | 0.17 | 1 | cimA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.; Tiwari, A.; Srivastava, S. A Genome-Scale Metabolic Model of Thalassiosira pseudonana CCMP 1335 for a Systems-Level Understanding of Its Metabolism and Biotechnological Potential. Microorganisms 2020, 8, 1396. https://doi.org/10.3390/microorganisms8091396
Ahmad A, Tiwari A, Srivastava S. A Genome-Scale Metabolic Model of Thalassiosira pseudonana CCMP 1335 for a Systems-Level Understanding of Its Metabolism and Biotechnological Potential. Microorganisms. 2020; 8(9):1396. https://doi.org/10.3390/microorganisms8091396
Chicago/Turabian StyleAhmad, Ahmad, Archana Tiwari, and Shireesh Srivastava. 2020. "A Genome-Scale Metabolic Model of Thalassiosira pseudonana CCMP 1335 for a Systems-Level Understanding of Its Metabolism and Biotechnological Potential" Microorganisms 8, no. 9: 1396. https://doi.org/10.3390/microorganisms8091396