
Clinical Application of Metagenomic Next-Generation Sequencing in Patients with Hematologic Malignancies Suffering from Sepsis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Next-Generation Sequencing Methodology
2.3. Bioinformatics
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, C.Y.; Tsay, W.; Tang, J.L.; Tien, H.F.; Chen, Y.C.; Chang, S.C. Epidemiology of bloodstream infections in patients with haematological malignancies with and without neutropenia. Epidemiol. Infec. 2010, 138, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Rosa, R.G.; Goldani, L.Z. Cohort study of the impact of time to antibiotic administration on mortality in patients with febrile neutropenia. Antimicrob. Agents Chemother. 2014, 58, 3799–3803. [Google Scholar] [CrossRef] [Green Version]
- Chamilos, G.; Lionakis, M.S.; Kontoyiannis, D.P. Call for Action: Invasive Fungal Infections Associated with Ibrutinib and Other Small Molecule Kinase Inhibitors Targeting Immune Signaling Pathways. Clin. Infect. Dis. 2018, 66, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Marchesi, F.; Pimpinelli, F.; Ensoli, F.; Mengarelli, A. Cytomegalovirus infection in hematologic malignancy settings other than the allogeneic transplant. Hematol. Oncol. 2018, 36, 381–391. [Google Scholar] [CrossRef]
- Moghnieh, R.; Estaitieh, N.; Mugharbil, A.; Jisr, T.; Abdallah, D.I.; Ziade, F.; Sinno, L.; Ibrahim, A. Third generation cephalosporin resistant Enterobacteriaceae and multidrug resistant gram-negative bacteria causing bacteremia in febrile neutropenia adult cancer patients in Lebanon, broad spectrum antibiotics use as a major risk factor, and correlation with poor prognosis. Front. Cell Infect. Microbiol. 2015, 5, 11. [Google Scholar] [PubMed]
- Snyder, M.; Pasikhova, Y.; Baluch, A. Early antimicrobial de-escalation and stewardship in adult hematopoietic stem cell transplantation recipients: Retrospective review. Open Forum Infect. Dis. 2017, 4, 226. [Google Scholar] [CrossRef]
- Miao, Q.; Ma, Y.; Wang, Q.; Pan, J.; Zhang, Y.; Jin, W.; Yao, Y.; Su, Y.; Huang, Y.; Wang, M.; et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin. Infect. Dis. 2018, 67, S231–S240. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Maio, Q.; Ma, Y.; Ling, Y.; Jin, W.; Su, Y.; Wang, Q.; Pan, J.; Zhang, J.; Cheng, H.; Yaun, J.; et al. Evaluation of superinfection, antimicrobial usage, and airway microbiome with metagenomic sequencing in COVID-19 patients: A cohort study in Shanghai. J. Microbiol. Immunol. Infect. 2020, in press. [Google Scholar]
- Li, H.; Gao, H.; Meng, H.; Wang, Q.; Li, S.; Chen, H.; Li, Y.; Wang, H. Detection of pulmonary infectious pathogens from lung biopsy tissues by metagenomic next-generation sequencing. Front. Cell. Infect. Microbiol. 2018, 8, 205. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Han, Y.; Feng, J. Metagenomic next-generation sequencing for mixed pulmonary infection diagnosis. BMC Pulm. Med. 2019, 19, 252. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.R.; O’Donovan, B.D.; Gelfand, J.M.; Sample, H.A.; Chow, F.C.; Betjemann, J.P.; Shah, M.; Richie, M.; Gorman, M.; Haj, R.; et al. Chronic meningitis investigated via metagenomic next-generation sequencing. JAMA Neurol. 2018, 75, 947–955. [Google Scholar] [CrossRef]
- Simner, P.J.; Miller, H.B.; Breitwieser, F.P.; Pinilla, M.G.; Pardo, C.A.; Salzberg, S.L.; Sears, C.; Thomas, D.; Eberhart, C.; Caroll, K. Development and optimization of metagenomic next-generation sequencing methods for cerebrospinal fluid diagnostics. J. Clin. Microbiol. 2018, 56, e00472-18. [Google Scholar] [CrossRef] [Green Version]
- Gyarmati, P.; Kjellander, C.; Aust, C.; Song, Y.; Öhrmalm, L.; Giske, C.G. Metagenomic analysis of bloodstream infections in patients with acute leukemia and therapy-induced neutropenia. Sci. Rep. 2016, 6, 23532. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernanrd, G.; Chiche, J.; Coopersmith, C.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Kunin, V.; Copeland, A.; Lapidus, A.; Mavromatis, K.; Hugenholtz, P. A bioinformatician’s guide to metagenomics. Microbiol. Mol. Biol. Rev. 2008, 72, 557–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Su, X.; Wang, A.; Xu, J.; Ning, K. QC-Chain: Fast and holistic quality control method for next-generation sequencing data. PLoS ONE 2013, 8, e60234. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Kryukov, K.; Nakagawa, S.; Takeuchi, J.S.; Takeshita, M.; Kirimura, Y.; Ishihara, I.; Aoki, H.; Inocoki, S.; Imanishi, C.; et al. Detection of pathogenic bacteria in the blood from sepsis patients using 16S rRNA gene amplicon sequencing analysis. PLoS ONE 2018, 13, e0202049. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, F.M.; Al-Amri, A.; Shaikh, S.S.; Muzaheed, A.A.I.; Acharya, S. Direct DNA sequencing-based analysis of microbiota associated with hematological malignancies in the Eastern Province of Saudi Arabia. Biomed. Res. Int. 2021, 3, 4202019. [Google Scholar]
- Fida, M.; Wolf, M.J.; Hamdi, A.; Vijayvargiya, P.; Esquer Garrigos, Z.; Khalil, S. Detection of pathogenic bacteria from septic patients using 16S ribosomal RNA gene-targeted metagenomic sequencing. Clin. Infect. Dis. 2021, 73, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ai, J.; Cui, P.; Zhu, Y.; Wu, H.; Zhang, W. Diagnostic value and clinical application of next-generation sequencing for infections in immunosuppressed patients with corticosteroid therapy. Ann. Transl. Med. 2020, 8, 227. [Google Scholar] [CrossRef]
- Rolston, K.V.; Bodey, G.P.; Safdar, A. Polymicrobial infection in patients with cancer: An underappreciated and underreported entity. Clin. Infect. Dis. 2007, 45, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, Y.; Li, T.; Rong, H.M.; Tong, Z.H. Diagnosis of Pneumocystis jirovecii pneumonia with serum cell-free DNA in non-HIV-infected immunocompromised patients. Oncotarget 2017, 8, 71946–71953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakil, E.; Evans, S.E. Viral pneumonia in patients with hematologic malignancy or hematopoietic stem cell transplantation. Clin. Chest. Med. 2017, 38, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.W.; Qu, J.L.; Wan, J.; Xu, Y.H.; Shan, Y.; Wu, L.X.; Zheng, J.; Jiang, W.; Chen, Q.; Zhu, Y.; et al. Effects of viral infection and microbial diversity on patients with sepsis: A retrospective study based on metagenomic next-generation sequencing. World J. Emerg. Med. 2021, 12, 29–35. [Google Scholar] [CrossRef]
- Popescu, C.M.; Ursache, A.L.; Feketea, G.; Bocsan, C.; Jimbu, L.; Mesaros, O.; Edwars, M.; Wang, H.; Berceanu, I.; Neaga, A.; et al. Are community acquired respiratory viral infections an underestimated burden in hematology patients? Microorganisms 2019, 7, 521. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.Y.; Wang, H.Y.; Zhou, Y.; Zhang, H.C.; Zhu, Y.M.; Zhou, X.; Ying, Y.; Cui, P.; Wu, H.; Zhang, W.; et al. Improving Pulmonary Infection Diagnosis with Metagenomic Next Generation Sequencing. Front. Cell. Infect. Microbiol. 2021, 10, 567615. [Google Scholar] [CrossRef] [PubMed]
- Grumaz, S.; Grumaz, C.; Vainshtein, Y.; Stevens, P.; Glanz, K.; Decker, S.O.; Hofer, S.; Weigand, M.; Brenner, H.; Sohn, K. Enhanced performance of next-generation sequencing diagnostics compared with standard of care microbiological diagnostics in patients suffering from septic shock. Crit. Care Med. 2019, 47, e394–e402. [Google Scholar] [CrossRef] [PubMed]
- Camargo, J.F.; Ahmed, A.A.; Lindner, M.S.; Morris, M.I.; Anjan, S.; Anderson, A.D.; Prado, C.; Dalai, S.; Martinez, O.; Komanduri, K. Next-generation sequencing of microbial cell-free DNA for rapid noninvasive diagnosis of infectious diseases in immunocompromised hosts. F1000Res 2019, 8, 1194. [Google Scholar] [CrossRef]
- Nikkari, S.; McLaughlin, I.J.; Bi, W.; Dodge, D.E.; Relman, D.A. Does blood of healthy subjects contain bacterial ribosomal DNA? J. Clin. Microbiol. 2001, 39, 1956–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosiewski, T.; Ludwig-Galezowska, A.H.; Huminska, K.; Sroka-Oleksiak, A.; Radkowski, P.; Salamon, D.; Bulanda, M.; Wolkow, M. Comprehensive detection and identification of bacterial DNA in the blood of patients with sepsis and healthy volunteers using next-generation sequencing method-the observation of DNAemia. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tusgul, S.; Prodhom, G.; Senn, L.; Meuli, R.; Bochud, P.Y.; Giulieri, S.G. Bacillus cereus bacteraemia: Comparison between haematologic and nonhaematologic patients. New Microbes New Infect. 2016, 15, 65–71. [Google Scholar] [CrossRef] [PubMed]
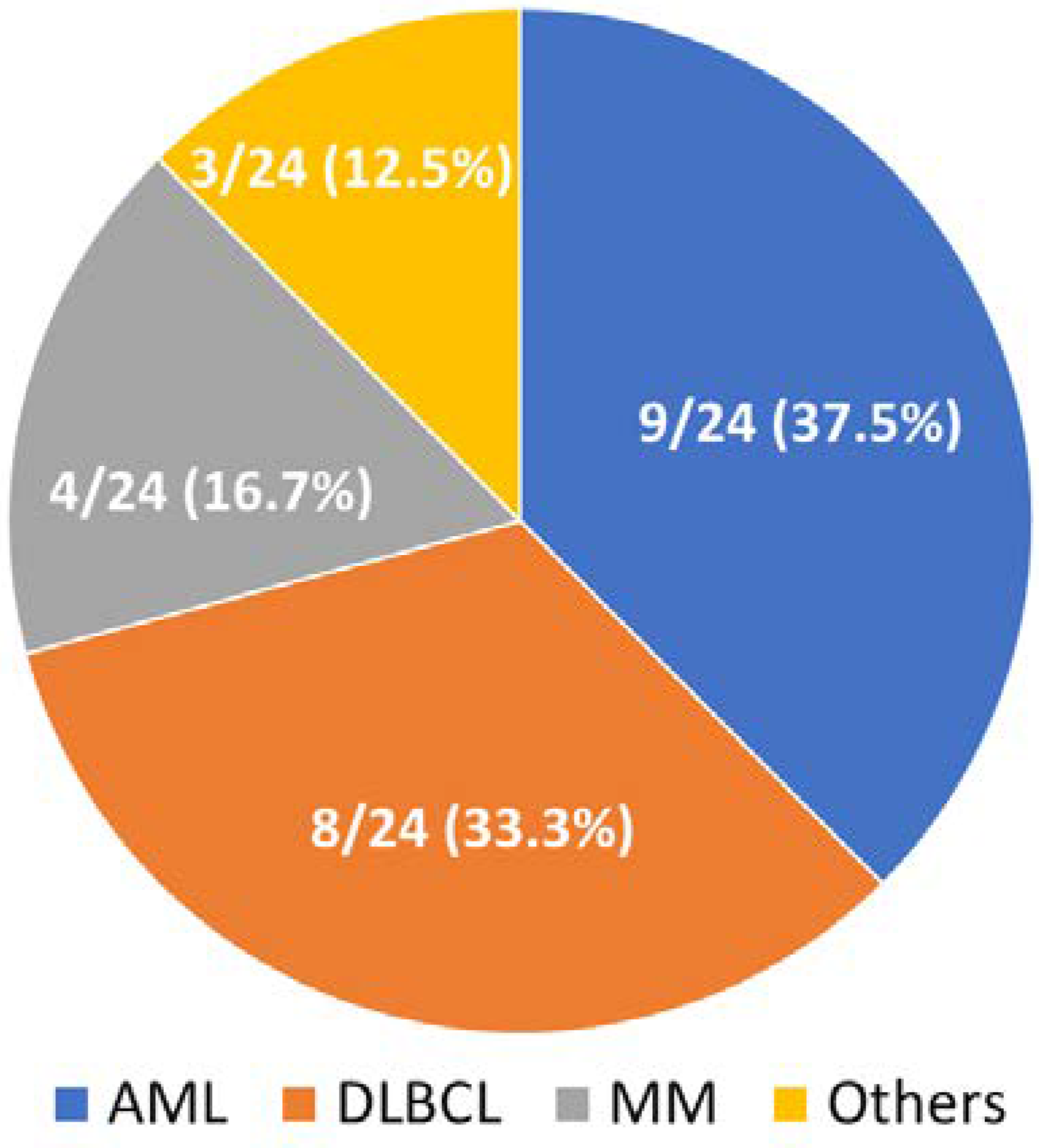

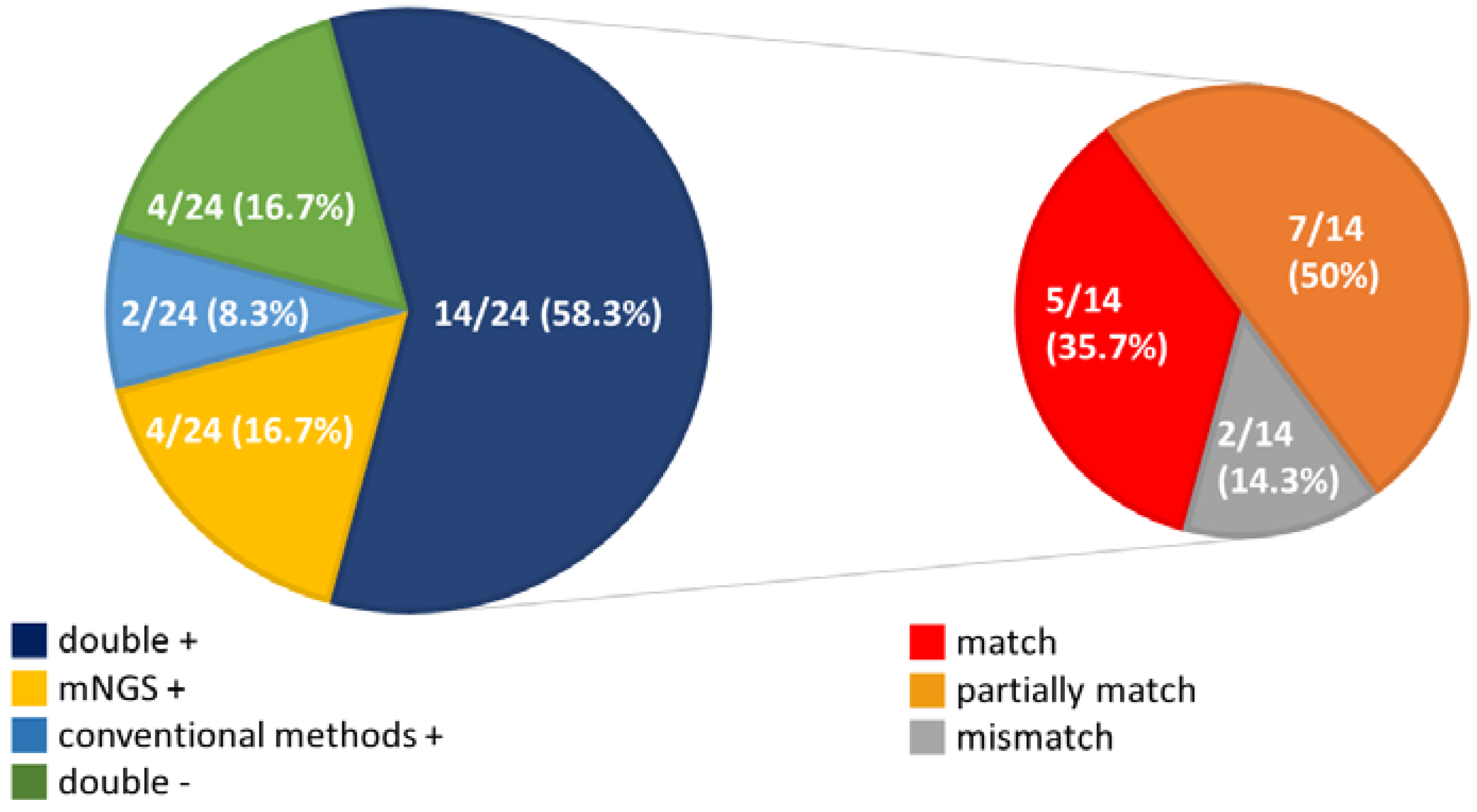

{kind=link}
{kind=link}
{kind=link}
| No. | Age/Gender | Underlying Diseases | Neutropenia | Clinical Diagnosis of Infection Disease | Blood Cultures | Additional Laboratory Tests | mNGS Results (Reads, Relative Abundance [%]) | Interval between Fever Onset and mNGS Sampling (Days) | mNGS Sampled before Effective Antimicrobial Agents |
|---|---|---|---|---|---|---|---|---|---|
| Information of conventional diagnostic method equal to mNGS (n = 9) | |||||||||
| 1 | 83/M | AML | Y | BSI | Bacillus cereus | B. cereus (100, 0.627) | 3 | Y | |
| 2 | 83/M | AML | Y | CAP | Pseudomona aeruginosa | P. aeruginosa (4022, 21.86) | 1 | Y | |
| 3 | 70/M | AML | Y | HAP | Klebsiella pneumoniae | K. pneumoniae (55, 0.45) | 1 | N | |
| 4 | 74/M | DLBCL | N | HAP | Serum and sputum Cytomegalovirus PCR (+) | CMV (2233, 10.63) | 1 | Y | |
| 5 | 66/M | MM | N | HAP | Serum and sputum CMV viral load: (+) Bronchial wash Pneumocystis jirovecii PCR (+) | CMV (19, 0.17) P. jirovecii (16, 0.14) | 1 | N | |
| 6 | 73/M | AML | N | IAI | 2 | - | |||
| 7 | 69/M | DLBCL | N | HLH | 3 | - | |||
| 8 | 80/M | AML | Y | HAP | 0 | - | |||
| 9 | 44/M | AML | Y | IAI | 1 | - | |||
| More information of mNGSs (n = 10) | |||||||||
| 10 | 64/F | DLBCL | N | UTI | K. pneumoniae | K. pneumoniae (46, 0.51) CMV (27, 0.3) | 1 | N | |
| 11 | 75/M | DLBCL | N | CRBSI | Enterobacter cloacae | E. cloacae (229, 1.93) Enterococcus faecium (26, 0.22) Acinetobacter baumannii (18, 0.15) | 0 | Y | |
| 12 | 76/M | AML | Y | VAP | Burkholderia cepacia complexE. faecium | Human mastadenovirus C (22,355, 79.62) E. faecium (47, 0.17) Bukholderia ubonensis (13, 0.05) | 0 | Y | |
| 13 | 61/F | T-LGLL | Y | IAI | Escherichia coli | Shigella dysenteriae (48, 0.09) K. pneumoniae (45, 0.08) E. cloacae (24, 0.04) Candida albicans (10, 0.02) | 0 | Y | |
| 14 | 53/M | MM | Y | IAI | K. pneumoniae | K. pneumoniae (113, 1.66) P. aeruginosa (18, 0.26) E. coli (5, 0.07) | 2 | Y | |
| 15 | 76/M | MM | N | UTI | E. coli | CMV (5260, 49.51) E.coli (33, 0.31) E. faecium (31, 0.29) | 4 | N | |
| 16 | 66/M | MM | Y | IAI | K. pneumoniae (210, 2.14) CMV (44, 0.34) C. albicans (4, 0.03) | 0 | Y | ||
| 17 | 55/M | AML | N | HSV-1 (42, 0.18) Cryptococcus neoformans (4, 0.02) | 2 | N | |||
| 18 | 62/M | DLBCL | N | HHV-6 (5310, 23.58) | 3 | N | |||
| 19 | 61/M | DLBCL | Y | HAP | CMV (14, 0.05) | 1 | N | ||
| More information of conventional methods (n = 3) | |||||||||
| 20 | 64/F | DLBCL | N | HAP | K. pneumoniae | Serum CMV viral load: (+) | CMV (39, 0.33) | 1 | N |
| 21 | 61/M | MCL | Y | IAI | Aeromonas veronii | 2 | N | ||
| 22 | 37/M | ALL | Y | HAP | E. coli | 1 | N | ||
| Inconsistent results between mNGS and conventional methods (n = 2) | |||||||||
| 23 | 82/M | DLBCL | Y | IAI | S. enterica E. faecium | C. albicans (310, 1.86) S. enterica (177, 1.06) CMV (46, 0.28) | 2 | N | |
| 24 | 80/F | AML | Y | BSI | Candia tropicalis | E. coli (5, 0.05) | 2 | N | |
| No. | Raw Reads | Clean Reads | Human Reads | Human (%) | Bacterial Reads | Bacterial (%) | Fungal Reads | Fungal (%) | Viral Reads | Viral (%) | Unclassified Reads | Unclassified (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 61,631,438 | 372,544 | 59,667,222 | 96.8 | 3874 | 0.006493 | 80 | 0.000134 | 16 | 0.000027 | 368,574 | 98.934354 |
| 2 | 47,018,822 | 751,060 | 45,392,646 | 96.5 | 6265 | 0.013802 | 549 | 0.001209 | 47 | 0.000104 | 744,199 | 99.086491 |
| 3 | 44,622,470 | 754,142 | 43,207,034 | 96.8 | 2199 | 0.005089 | 585 | 0.001354 | 32 | 0.000074 | 751,326 | 99.626596 |
| 4 | 61,310,234 | 1,295,834 | 58,817,288 | 95.9 | 2511 | 0.004269 | 889 | 0.001511 | 2293 | 0.003899 | 1,290,141 | 99.560669 |
| 5 | 47,892,512 | 484,488 | 46,624,618 | 97.4 | 1709 | 0.003665 | 297 | 0.000637 | 37 | 0.000079 | 482,445 | 99.578318 |
| 6 | 140,925,496 | 1,038,042 | 136,530,428 | 96.9 | 4472 | 0.003275 | 504 | 0.000369 | 18 | 0.000013 | 1,033,048 | 99.518902 |
| 7 | 48,307,008 | 370,708 | 46,691,368 | 96.7 | 6860 | 0.014692 | 168 | 0.000360 | 81 | 0.000173 | 363,599 | 98.082318 |
| 8 | 34,052,126 | 255,536 | 33,138,968 | 97.3 | 2673 | 0.008066 | 93 | 0.000281 | 11 | 0.000033 | 252,759 | 98.913265 |
| 9 | 22,177,606 | 485,240 | 21,191,972 | 95.6 | 33,501 | 0.158083 | 325 | 0.001534 | 223 | 0.001052 | 451,191 | 92.983060 |
| 10 | 37,286,458 | 413,648 | 36,212,734 | 97.1 | 1441 | 0.003979 | 260 | 0.000718 | 89 | 0.000246 | 411,858 | 99.567265 |
| 11 | 89,094,784 | 506,686 | 85,454,082 | 95.9 | 1976 | 0.002312 | 98 | 0.000115 | 438 | 0.000513 | 504,174 | 99.504229 |
| 12 | 20,962,736 | 348,342 | 20,138,688 | 96.1 | 1644 | 0.008163 | 268 | 0.001331 | 5289 | 0.026263 | 341,141 | 97.932779 |
| 13 | 31,182,134 | 912,828 | 29,495,118 | 94.6 | 62,320 | 0.211289 | 594 | 0.002014 | 471 | 0.001597 | 849,443 | 93.056195 |
| 14 | 61,919,982 | 393,804 | 58,928,694 | 95.2 | 551 | 0.000935 | 96 | 0.000163 | 1 | 0.000002 | 393,156 | 99.835451 |
| 15 | 20,962,736 | 348,342 | 20,138,688 | 96.1 | 1644 | 0.008163 | 268 | 0.001331 | 5289 | 0.026263 | 341,141 | 97.932779 |
| 16 | 35,937,874 | 418,856 | 34,893,316 | 97.1 | 5419 | 0.015530 | 227 | 0.000651 | 79 | 0.000226 | 413,131 | 98.633182 |
| 17 | 167,008,212 | 1,087,152 | 161,133,136 | 96.5 | 4440 | 0.002755 | 309 | 0.000192 | 61 | 0.000038 | 1,082,342 | 99.557560 |
| 18 | 70,752,128 | 523,784 | 68,587,748 | 96.9 | 8907 | 0.012986 | 227 | 0.000331 | 5409 | 0.007886 | 509,241 | 97.223474 |
| 19 | 44,666,980 | 754,222 | 42,965,016 | 96.2 | 30,655 | 0.071349 | 521 | 0.001213 | 78 | 0.000182 | 722,968 | 95.856127 |
| 20 | 35,576,312 | 367,184 | 34,538,460 | 97.1 | 4342 | 0.012571 | 236 | 0.000683 | 67 | 0.000194 | 362,539 | 98.734967 |
| 21 | 87,638,198 | 467,968 | 83,603,160 | 95.4 | 2608 | 0.003119 | 53 | 0.000063 | 15 | 0.000018 | 465,292 | 99.428166 |
| 22 | 81,554,252 | 421,902 | 78,379,234 | 96.1 | 3041 | 0.003880 | 73 | 0.000093 | 4 | 0.000005 | 418,784 | 99.260966 |
| 23 | 71,295,070 | 479,434 | 68,314,830 | 95.8 | 4688 | 0.006862 | 477 | 0.000698 | 75 | 0.000110 | 474,194 | 98.907045 |
| 24 | 54,129,834 | 467,204 | 52,886,676 | 97.7 | 736 | 0.001392 | 264 | 0.000499 | 17 | 0.000032 | 466,187 | 99.782322 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.-D.; Yen, T.-Y.; Liu, P.-Y.; Wu, U.-I.; Bhan, P.; Li, Y.-C.; Chi, C.-H.; Sheng, W.-H. Clinical Application of Metagenomic Next-Generation Sequencing in Patients with Hematologic Malignancies Suffering from Sepsis. Microorganisms 2021, 9, 2309. https://doi.org/10.3390/microorganisms9112309
Liu W-D, Yen T-Y, Liu P-Y, Wu U-I, Bhan P, Li Y-C, Chi C-H, Sheng W-H. Clinical Application of Metagenomic Next-Generation Sequencing in Patients with Hematologic Malignancies Suffering from Sepsis. Microorganisms. 2021; 9(11):2309. https://doi.org/10.3390/microorganisms9112309
Chicago/Turabian StyleLiu, Wang-Da, Ting-Yu Yen, Po-Yo Liu, Un-In Wu, Prerana Bhan, Yu-Chi Li, Chih-Hung Chi, and Wang-Huei Sheng. 2021. "Clinical Application of Metagenomic Next-Generation Sequencing in Patients with Hematologic Malignancies Suffering from Sepsis" Microorganisms 9, no. 11: 2309. https://doi.org/10.3390/microorganisms9112309