
Peat-Inhabiting Verrucomicrobia of the Order Methylacidiphilales Do Not Possess Methanotrophic Capabilities

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of Verrucomicrobia Diversity in Ten Different Peatlands of European North Russia
2.2. Peat Sampling for Incubation Studies
2.3. Determination of CH4 Oxidation Activity of Peat Samples and Incubation Experiments
2.4. 16S rRNA Gene Sequencing and Analysis
2.5. Sequencing of Metagenomic DNA and Assembly of MAGs
2.6. Genome Annotation and Analysis
3. Results and Discussion
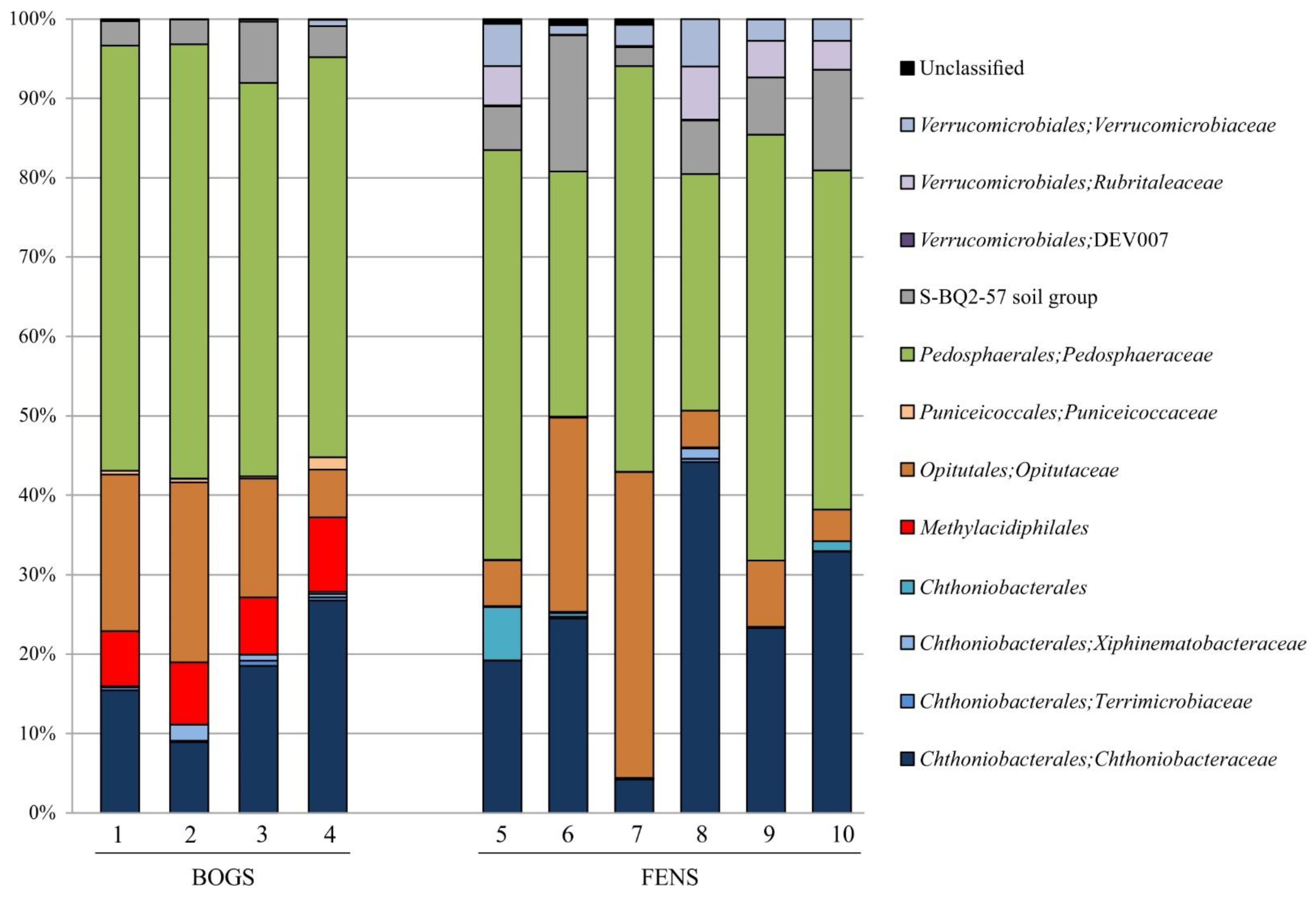
3.1. Verrucomicrobia Diversity Patterns in Peatlands
3.2. Most Abundant OTUs of Methylacidiphilales
3.3. Methane-Induced Shifts in the Microbial Community Structure
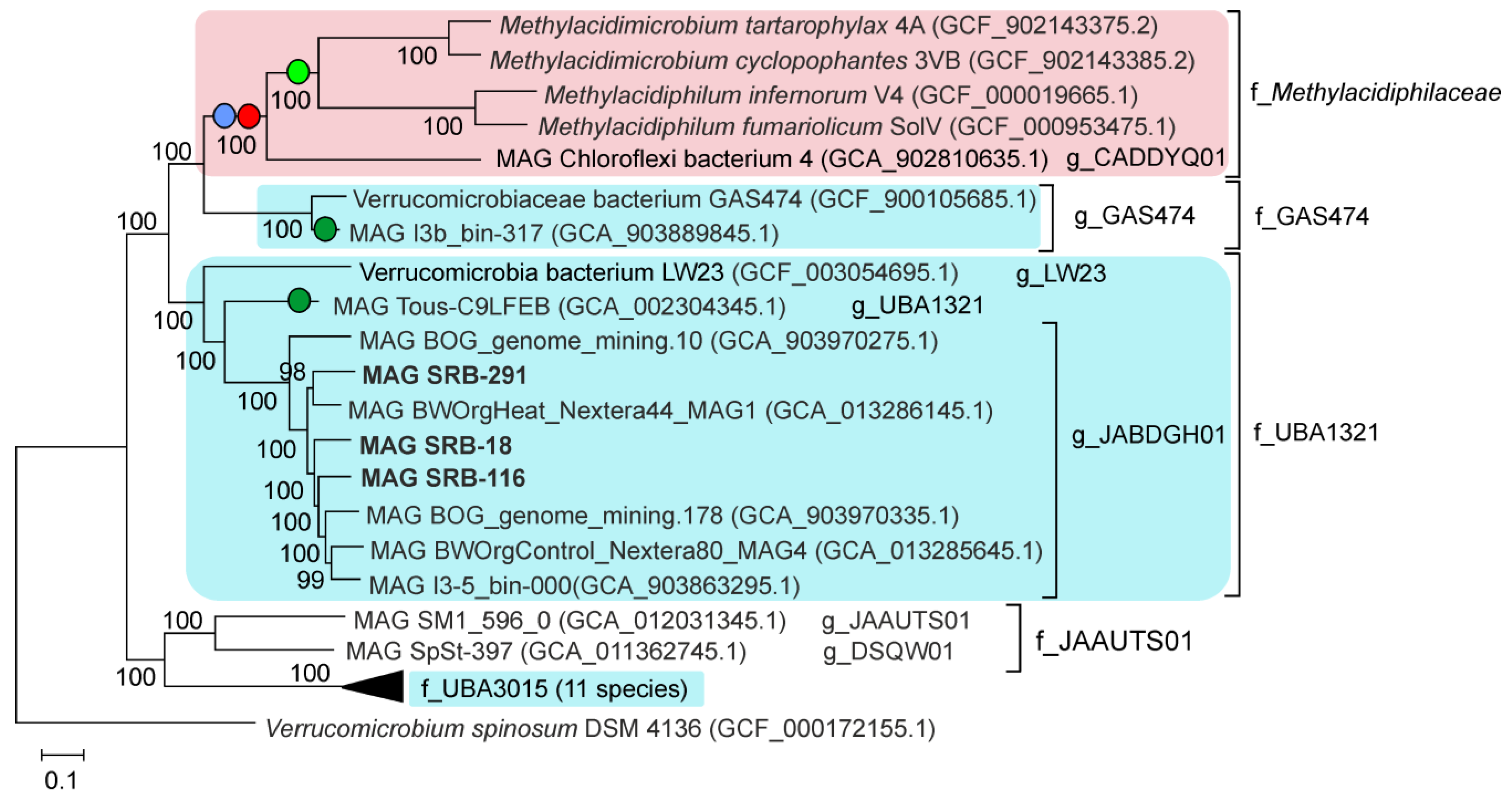
3.4. Assembly and Phylogenetic Placement of MAGs of Methylacidiphilales
3.5. Genome-Based Metabolic Predictions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunfield, P.F.; Yuryev, A.; Senin, P.; Smirnova, A.V.; Stott, M.B.; Hou, S.; Ly, B.; Saw, J.H.; Zhou, Z.; Ren, Y.; et al. Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 2007, 450, 879–882. [Google Scholar] [CrossRef]
- Pol, A.; Heijmans, K.; Harhangi, H.R.; Tedesco, D.; Jetten, M.S.M.; Op Den Camp, H.J.M. Methanotrophy below pH 1 by a new Verrucomicrobia species. Nature 2007, 450, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Jensen, S.; Reigstad, L.J.; Larsen, Ø.; Birkeland, N.K. Methane oxidation at 55 °C and pH 2 by a thermoacidophilic bacterium belonging to the Verrucomicrobia phylum. Proc. Natl. Acad. Sci. USA 2008, 105, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Op den Camp, H.J.M.; Islam, T.; Stott, M.B.; Harhangi, H.R.; Hynes, A.; Schouten, S.; Jetten, M.S.M.; Birkeland, N.K.; Pol, A.; Dunfield, P.F. Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ. Microbiol. Rep. 2009, 1, 293–306. [Google Scholar] [CrossRef] [PubMed]
- van Teeseling, M.; Pol, A.; Harhangi, H.; van der Zwart, S.; Jetten, M.; Op den Camp, H.; van Niftrik, L. Expanding the verrucomicrobial methanotrophic world: Description of three novel species of Methylacidimicrobium gen. nov. Appl. Environ. Microbiol. 2014, 80, 6782–6791. [Google Scholar] [CrossRef] [Green Version]
- Khadem, A.F.; Pol, A.; Wieczorek, A.; Mohammadi, S.S.; Francoijs, K.J.; Stunnenberg, H.G.; Jetten, M.S.M.; Op den Camp, H.J.M. Autotrophic methanotrophy in verrucomicrobia: Methylacidiphilum fumariolicum SolV uses the Calvin-Benson-Bassham cycle for carbon dioxide fixation. J. Bacteriol. 2011, 193, 4438–4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pol, A.; Barends, T.R.M.; Dietl, A.; Khadem, A.F.; Eygensteyn, J.; Jetten, M.S.M.; Op den Camp, H.J.M. Rare earth metals are essential for methanotrophic life in volcanic mudpots. Environ. Microbiol. 2014, 16, 255–264. [Google Scholar] [CrossRef]
- Carere, C.R.; Hards, K.; Houghton, K.M.; Power, J.F.; McDonald, B.; Collet, C.; Gapes, D.J.; Sparling, R.; Boyd, E.S.; Cook, G.M.; et al. Mixotrophy drives niche expansion of verrucomicrobial methanotrophs. ISME J. 2017, 11, 2599–2610. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.A.; Peeters, S.H.; Versantvoort, W.; Picone, N.; Pol, A.; Jetten, M.S.M.; Op Den Camp, H.J.M. Verrucomicrobial methanotrophs: Ecophysiology of metabolically versatile acidophiles. FEMS Microbiol. Rev. 2021, 45, fuab007. [Google Scholar] [CrossRef]
- Pagaling, E.; Yang, K.; Yan, T. Pyrosequencing reveals correlations between extremely acidophilic bacterial communities with hydrogen sulphide concentrations, pH and inert polymer coatings at concrete sewer crown surfaces. J. Appl. Microbiol. 2014, 117, 50–64. [Google Scholar] [CrossRef] [Green Version]
- Sharp, C.E.; Smirnova, A.V.; Graham, J.M.; Stott, M.B.; Khadka, R.; Moore, T.R.; Grasby, S.E.; Strack, M.; Dunfield, P.F. Distribution and diversity of Verrucomicrobia methanotrophs in geothermal and acidic environments. Environ. Microbiol. 2014, 16, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Serkebaeva, Y.M.; Kim, Y.; Liesack, W.; Dedysh, S.N. Pyrosequencing-based assessment of the bacteria diversity in surface and subsurface peat layers of a northern wetland, with focus on poorly studied phyla and candidate divisions. PLoS ONE 2013, 8, e63994. [Google Scholar] [CrossRef] [Green Version]
- Putkinen, A.; Larmola, T.; Tuomivirta, T.; Siljanen, H.M.P.; Bodrossy, L.; Tuittila, E.S.; Fritze, H. Peatland succession induces a shift in the community composition of Sphagnum-associated active methanotrophs. FEMS Microbiol. Ecol. 2014, 88, 596–611. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, A.A.; Beletsky, A.V.; Rakitin, A.L.; Kadnikov, V.V.; Philippov, D.A.; Mardanov, A.V.; Ravin, N.V.; Dedysh, S.N. Closely located but totally distinct: Highly contrasting prokaryotic diversity patterns in raised bogs and eutrophic fens. Microorganisms 2020, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Dedysh, S.N.; Beletsky, A.V.; Ivanova, A.A.; Kulichevskaya, I.S.; Suzina, N.E.; Philippov, D.A.; Rakitin, A.L.; Mardanov, A.V.; Ravin, N.V. Wide distribution of Phycisphaera-like planctomycetes from WD2101 soil group in peatlands and genome analysis of the first cultivated representative. Environ. Microbiol. 2020, 23, 1510–1526. [Google Scholar] [CrossRef]
- Caporaso, J.; Kuczynski, J.; Stombaugh, J. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rime, T.; Phillips, M.; Stierli, B.; Hajdas, I.; Widmer, F.; Hartmann, M. Microbial diversity in European alpine permafrost and active layers. FEMS Microbiol. Ecol. 2016, 92, fiw018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 2015, e1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-R., L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Bünger, W.; Jiang, X.; Müller, J.; Hurek, T.; Reinhold-Hurek, B. Novel cultivated endophytic Verrucomicrobia reveal deep-rooting traits of bacteria to associate with plants. Sci. Rep. 2020, 10, 8692. [Google Scholar] [CrossRef] [PubMed]
- Dedysh, S.N. Exploring methanotroph diversity in acidic northern wetlands: Molecular and cultivation-based studies. Microbiology 2009, 78, 655–669. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, C.; Rodriguez-R., L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Anvar, S.Y.; Frank, J.; Pol, A.; Schmitz, A.; Kraaijeveld, K.; den Dunnen, J.T.; Op den Camp, H.J.M. The genomic landscape of the verrucomicrobial methanotroph Methylacidiphilum fumariolicum SolV. BMC Genomics 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravin, N.V.; Rakitin, A.L.; Ivanova, A.A.; Beletsky, A.V.; Kulichevskaya, I.S.; Mardanov, A.V.; Dedysh, S.N. Genome analysis of Fimbriiglobus ruber SP5T, a planctomycete with confirmed chitinolytic capability. Appl. Environ. Microbiol. 2018, 84, AEM.02645-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scigelova, M.; Crout, D.H.G. Microbial beta-N-acetylhexosaminidases and their biotechnological applications. Enzyme Microb. Technol. 1999, 25, 3–14. [Google Scholar] [CrossRef]
- Boronat, A.; Aguilar, J. Metabolism of L-fucose and L-rhamnose in Escherichia coli: Differences in induction of propanediol oxidoreductase. J. Bacteriol. 1981, 147, 181. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, C.; Sinha, S.; Chun, S.; Yeates, T.O.; Bobik, T.A. Diverse bacterial microcompartment organelles. Microbiol. Mol. Biol. Rev. 2014, 78, 438–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbilgin, O.; McDonald, K.L.; Kerfeld, C.A. Characterization of a planctomycetal organelle: A novel bacterial microcompartment for the aerobic degradation of plant saccharides. Appl. Environ. Microbiol. 2014, 80, 2193–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barns, S.M.; Takala, S.L.; Kuske, C.R. Wide distribution and diversity of members of the bacterial kingdom Acidobacterium in the environment. Appl. Environ. Microbiol. 1999, 65, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mires | Characteristics | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Coordinates | pH | Total Organic C (%) | N Total (%) | Sulfate (mg/L) | Fe (ppm) | Ca (ppm) | Mg (ppm) | P (ppm) | ||
| RAISED BOGS * | 1 | 59°56′56″ N 41°16′59″ E | 4.3 | 88.5 | 0.605 | 172 | 343 | 3522 | 634 | 614 |
| 2 | 60°46′29″ N 36°49′35″ E | 3.7 | 85.1 | 0.923 | 220 | 1347 | 4190 | 682 | 791 | |
| 3 | 59°27′10″ N 40°30′45″ E | 4.3 | 88 | 0.685 | 211 | 662 | 4191 | 905 | 721 | |
| 4 | 59°22′33″ N 39°59′26″ E | 4.1 | 81.5 | 1.16 | 200 | 5306 | 3765 | 816 | 1020 | |
| FENS ** | 1 | 59°56′31″ N 41°15′53″ E | 7.4 | 73.6 | 2.31 | 202 | 9387 | 29,834 | 2575 | 1179 |
| 2 | 60°46′08″ N 36°49′30″ E | 6.9 | 71.6 | 1.65 | 222 | 16,344 | 27,373 | 1078 | 1305 | |
| 3 | 59°47′08″ N 37°52′08″ E | 7.6 | 41.8 | 1.06 | 186 | 106,966 | 32,196 | 1599 | 8920 | |
| 4 | 61°08′18″ N 36°33′27″ E | 6.9 | 83.2 | 2.55 | 230 | 3455 | 15,968 | 2583 | 1049 | |
| 5 | 61°07′16″ N 36°33′21″ E | 6.5 | 48.6 | 1.51 | 607 | 19,264 | 8494 | 2665 | 1192 | |
| 6 | 60°30′42″ N 38°38′59″ E | 7.1 | 66.2 | 2.4 | 188 | 5333 | 31,193 | 2695 | 985 | |
| MAG ID | MAG Size (bp) | Completeness (%) | Redundancy (%) | No. of Contigs | Sequencing Coverage | No. of tRNA Genes | No. of Protein-Coding Genes |
|---|---|---|---|---|---|---|---|
| SRB-18 | 2,871,672 | 90.41 | 3.04 | 88 | 19.7 | 35 | 2843 |
| SRB-116 | 3,197,352 | 92.57 | 8.62 | 93 | 42.3 | 39 | 3241 |
| SRB-291 | 3,829,391 | 95.11 | 5.6 | 169 | 9.2 | 49 | 3864 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dedysh, S.N.; Beletsky, A.V.; Ivanova, A.A.; Danilova, O.V.; Begmatov, S.; Kulichevskaya, I.S.; Mardanov, A.V.; Ravin, N.V. Peat-Inhabiting Verrucomicrobia of the Order Methylacidiphilales Do Not Possess Methanotrophic Capabilities. Microorganisms 2021, 9, 2566. https://doi.org/10.3390/microorganisms9122566
Dedysh SN, Beletsky AV, Ivanova AA, Danilova OV, Begmatov S, Kulichevskaya IS, Mardanov AV, Ravin NV. Peat-Inhabiting Verrucomicrobia of the Order Methylacidiphilales Do Not Possess Methanotrophic Capabilities. Microorganisms. 2021; 9(12):2566. https://doi.org/10.3390/microorganisms9122566
Chicago/Turabian StyleDedysh, Svetlana N., Alexey V. Beletsky, Anastasia A. Ivanova, Olga V. Danilova, Shahjahon Begmatov, Irina S. Kulichevskaya, Andrey V. Mardanov, and Nikolai V. Ravin. 2021. "Peat-Inhabiting Verrucomicrobia of the Order Methylacidiphilales Do Not Possess Methanotrophic Capabilities" Microorganisms 9, no. 12: 2566. https://doi.org/10.3390/microorganisms9122566