
Escherichia coli Causing Recurrent Urinary Tract Infections: Comparison to Non-Recurrent Isolates and Genomic Adaptation in Recurrent Infections

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Bacterial Isolates
2.3. Sequencing, Assembly and Annotation
2.4. Phylogeny and Pairwise SNPs
2.5. Accessory Genome Content and Typing
2.6. Ethical Approval
3. Results and Discussion
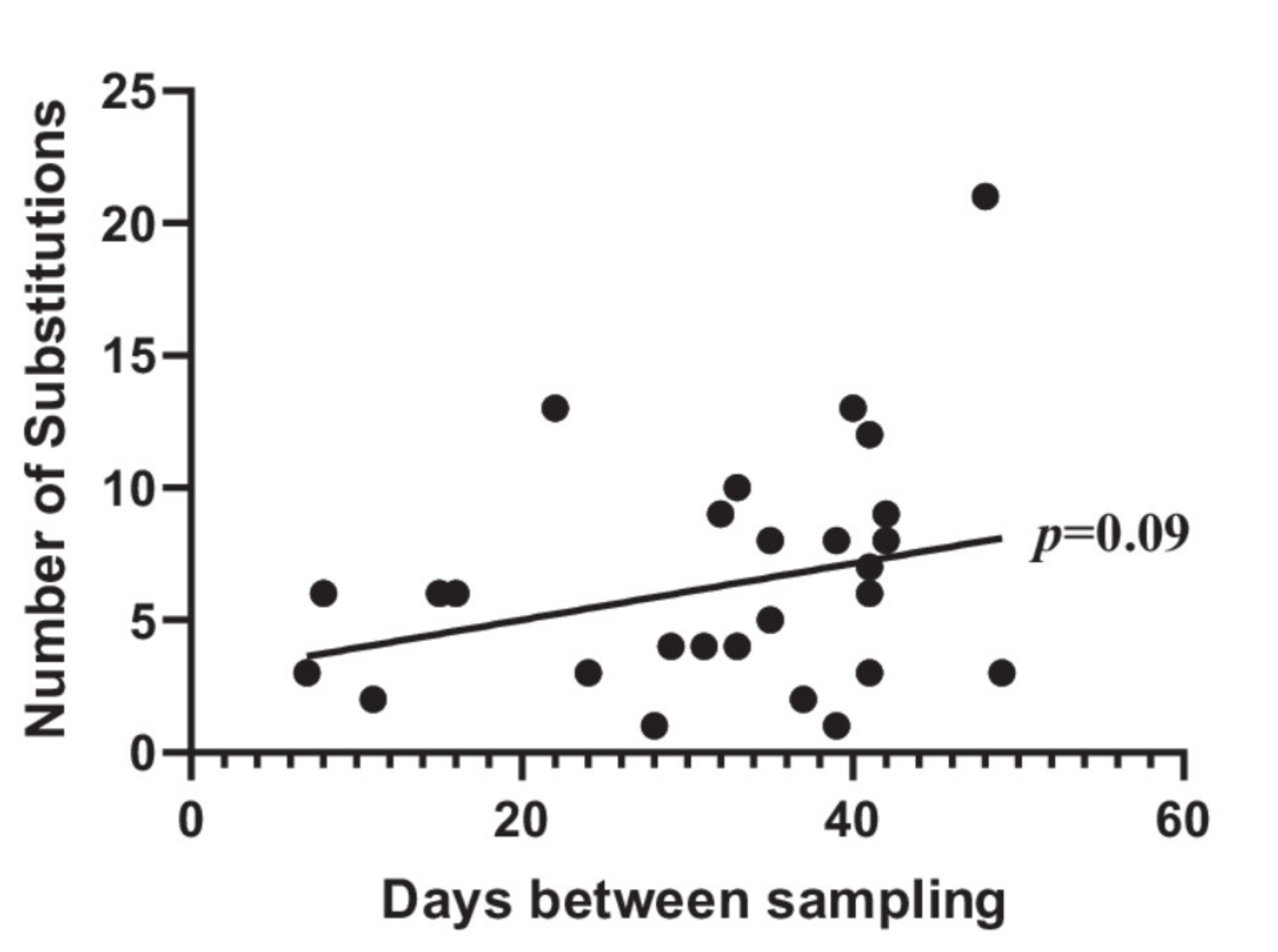
3.1. Adaptation between First and Second UTI
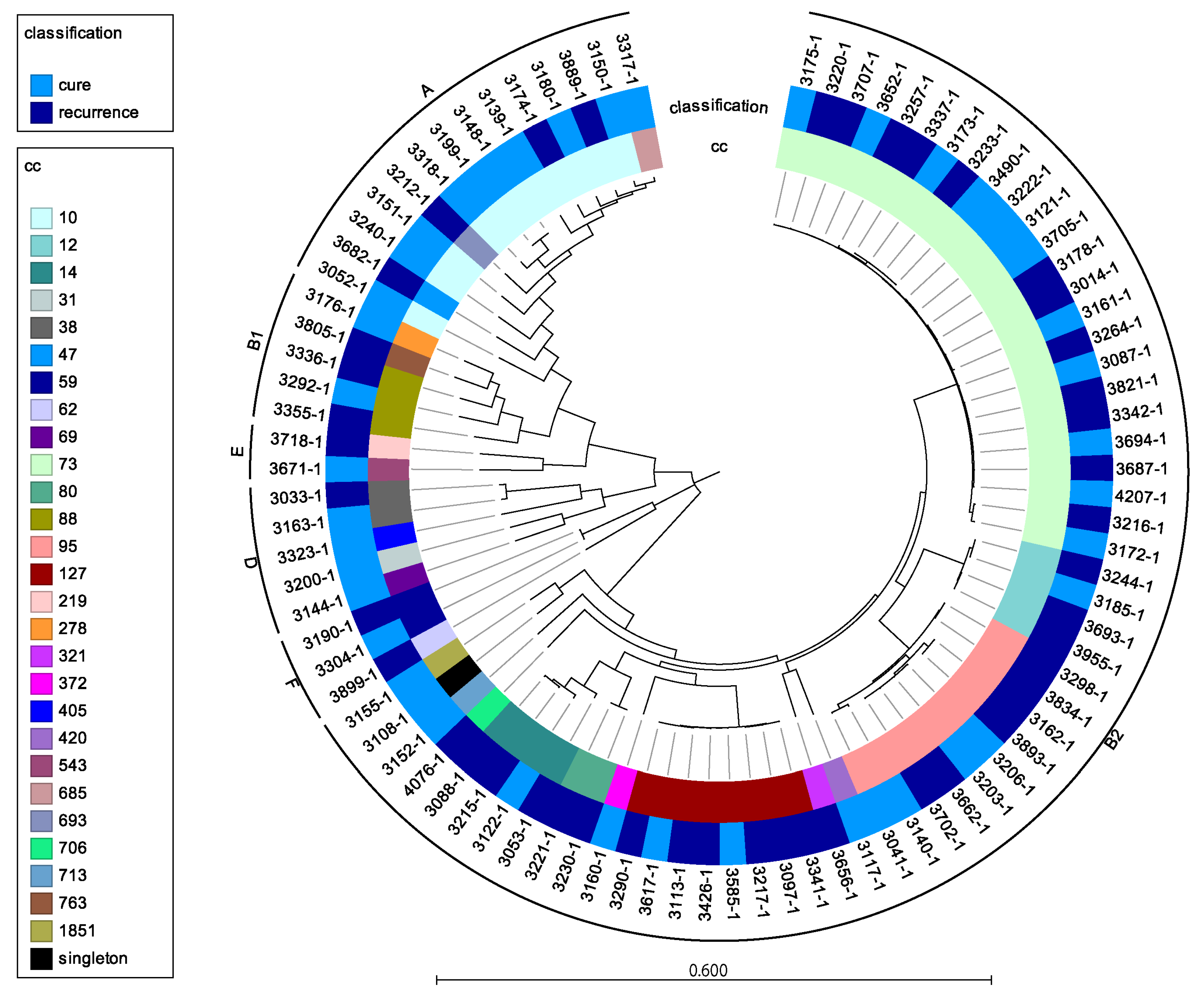
3.2. Do rUTI Isolates Differ Genetically from Non-rUTI Isolates?
3.3. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mabeck, C.E. Treatment of uncomplicated urinary tract infection in non-pregnant women. Postgrad. Med. J. 1972, 48, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Russo, T.A.; Stapleton, A.; Wenderoth, S.; Hooton, T.M.; Stamm, W.E. Chromosomal restriction fragment length polymorphism analysis of Escherichia coli strains causing recurrent urinary tract infections in young women. J. Infect. Dis. 1995, 172, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Hooton, T.M. Recurrent urinary tract infection in women. Int. J. Antimicrob. Agents 2001, 17, 259–268. [Google Scholar] [CrossRef]
- Thänert, R.; Reske, K.A.; Hink, T.; Wallace, M.A.; Wang, B.; Schwartz, D.J.; Seiler, S.; Cass, C.; Burnham, C.-A.D.; Dubberke, E.R.; et al. Comparative Genomics of Antibiotic-Resistant Uropathogens Implicates Three Routes for Recurrence of Urinary Tract Infections. MBio 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Forde, B.M.; Roberts, L.W.; Phan, M.-D.; Peters, K.M.; Fleming, B.A.; Russell, C.W.; Lenherr, S.M.; Myers, J.B.; Barker, A.P.; Fisher, M.A.; et al. Population dynamics of an Escherichia coli ST131 lineage during recurrent urinary tract infection. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Foxman, B.; Frerichs, R.R. Epidemiology of urinary tract infection: II. Diet, clothing, and urination habits. Am. J. Public Health 1985, 75, 1308–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karami, N.; Lindblom, A.; Yazdanshenas, S.; Lindén, V.; Åhrén, C. Recurrence of urinary tract infections with extended-spectrum β-lactamase-producing Escherichia coli caused by homologous strains among which clone ST131-O25b is dominant. J. Glob. Antimicrob. Resist. 2020, 22, 126–132. [Google Scholar] [CrossRef]
- Kärkkäinen, U.M.; Ikäheimo, R.; Katila, M.L.; Siitonen, A. Recurrence of urinary tract infections in adult patients with community-acquired pyelonephritis caused by E. coli: A 1-year follow-up. Scand. J. Infect. Dis. 2000, 32, 495–499. [Google Scholar] [CrossRef]
- Li, D.; Reid, C.J.; Kudinha, T.; Jarocki, V.M.; Djordjevic, S.P. Genomic analysis of trimethoprim-resistant extraintestinal pathogenic Escherichia coli and recurrent urinary tract infections. Microb. Genom. 2020, 6, 1–19. [Google Scholar] [CrossRef]
- Soto, S.M.; Smithson, A.; Martinez, J.A.; Horcajada, J.P.; Mensa, J.; Vila, J. Biofilm formation in uropathogenic Escherichia coli strains: Relationship with prostatitis, urovirulence factors and antimicrobial resistance. J. Urol. 2007, 177, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; O’Bryan, T.T.; Delavari, P.; Kuskowski, M.; Stapleton, A.; Carlino, U.; Russo, T. A Clonal relationships and extended virulence genotypes among Escherichia coli isolates from women with a first or recurrent episode of cystitis. J. Infect. Dis. 2001, 183, 1508–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, S.M.; Smithson, A.; Horcajada, J.P.; Martinez, J.A.; Mensa, J.P.; Vila, J. Implication of biofilm formation in the persistence of urinary tract infection caused by uropathogenic Escherichia coli. Clin. Microbiol. Infect. 2006, 12, 1034–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.L.; Wu, M.; Henderson, J.P.; Hooton, T.M.; Hibbing, M.E.; Hultgren, S.J.; Gordon, J.I. Genomic Diversity and Fitness of E. coli Strains Recovered from the Intestinal and Urinary Tracts of Women with Recurrent Urinary Tract Infection. Sci. Transl. Med. 2013, 5, 184ra60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, K.L.; Stegger, M.; Godfrey, P.A.; Feldgarden, M.; Andersen, P.S.; Frimodt-Møller, N. Adaptation of Escherichia coli traversing from the faecal environment to the urinary tract. Int. J. Med. Microbiol. 2016, 306, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Ferry, S.A.; Holm, S.E.; Stenlund, H.; Lundholm, R.; Monsen, T.J. Clinical and bacteriological outcome of different doses and duration of pivmecillinam compared with placebo therapy of uncomplicated lower urinary tract infection in women: The LUTIW project. Scand. J. Prim. Health Care 2007, 25, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejrnaes, K.; Sandvang, D.; Lundgren, B.; Ferry, S.; Holm, S.; Monsen, T.; Lundholm, R.; Frimodt-moller, N. Pulsed-Field Gel Electrophoresis Typing of Escherichia coli Strains from Samples Collected before and after Pivmecillinam or Placebo Treatment of Uncomplicated Community-Acquired Urinary Tract Infection in Women. J. Clin. Microbiol. 2006, 44, 1776–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, Y.H.; Lipsitch, M.; Feldgarden, M.; Arachchi, H.M.; Cerqueira, G.C.; Fitzgerald, M.; Godfrey, P.; Haas, B.J.; Murphy, C.I.; Russ, C.; et al. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc. Natl. Acad. Sci. USA 2012, 109, 3065–3070. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R. Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinform. 2010, 31, 11–5. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2017, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, O.; Christenson, J.K.; Denamur, E.; Gordon, D.M. The Clermont Escherichia coli phylo-typing method revisited: Improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013, 5, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A. Metabolic Networks and Their Evolution. In Evolutionary Systems Biology; Springer: New York, NY, USA, 2012; Volume 751, pp. 39–40. [Google Scholar] [CrossRef]
- Cavicchioli, R.; Schroder, I.; Constanti, M.; Gunsalus, R.P. The NarX and NarQ sensor-transmitter proteins of Escherichia coli each require two conserved histidines for nitrate-dependent signal transduction to NarL. J. Bacteriol. 1995, 177, 2416–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Garcia, F.; Ruiz-Perez, F.; Cataldi, Á.; Larzábal, M. Type VI Secretion System in Pathogenic Escherichia coli: Structure, Role in Virulence, and Acquisition. Front. Microbiol. 2019, 10, 1965. [Google Scholar] [CrossRef] [Green Version]
- Spurbeck, R.R.; Stapleton, A.E.; Johnson, J.R.; Walk, S.T.; Hooton, T.M.; Mobley, H.L.T. Fimbrial profiles predict virulence of uropathogenic Escherichia coli strains: Contribution of ygi and yad fimbriae. Infect. Immun. 2011, 79, 4753–4763. [Google Scholar] [CrossRef] [Green Version]
- Lobato-Márquez, D.; Díaz-Orejas, R.; García-del Portillo, F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef] [Green Version]
- Lasaro, M.A.; Salinger, N.; Zhang, J.; Wang, Y.; Zhong, Z.; Goulian, M.; Zhu, J. F1C fimbriae play an important role in biofilm formation and intestinal colonization by the Escherichia coli commensal strain Nissle 1917. Appl. Environ. Microbiol. 2009, 75, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.J.; Seed, P.C.; Hultgren, S.J. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell. Microbiol. 2007, 9, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.L.; Stegger, M.; Kiil, K.; Godfrey, P.A.; Feldgarden, M.; Lilje, B.; Andersen, P.S.; Frimodt-Møller, N. Whole-genome comparison of urinary pathogenic Escherichia coli and faecal isolates of UTI patients and healthy controls. Int. J. Med. Microbiol. 2017, 307, 497–507. [Google Scholar] [CrossRef]
- Stegger, M.; Leihof, R.F.; Baig, S.; Sieber, R.N.; Thingholm, K.R.; Marvig, R.L.; Frimodt-Møller, N.; Nielsen, K.L. A snapshot of diversity: Intraclonal variation of Escherichia coli clones as commensals and pathogens. Int. J. Med. Microbiol. 2020, 310, 151401. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene Annotation | Function | p-Value | p-Value FDR | Cure n (%) | rUTI n (%) | Isolate_Locus Tag |
|---|---|---|---|---|---|---|
| Overrepresented in rUTI isolates | ||||||
| Fimbria-related genes | ||||||
| papB | S/F1C fimbrial transcriptional regulator | 0.0062 | 0.619 | 14 (33) | 28 (62) | 3244-2_K_03164 |
| sfaH | S-fimbrial protein subunit | 0.0072 | 0.619 | 2 (5) | 12 (27) | 3244-2_K_03157 |
| mprA | Transcriptional repressor | 0.0072 | 0.619 | 26 (60) | 39 (87) | 3244-2_K_03155 |
| sfaF | S/F1C fimbrial biogenesis usher protein | 0.0118 | 0.619 | 15 (35) | 28(62) | 3244-2_K_03160 |
| sfaE | S/F1C fimbrial biogenesis chaperone | 0.0118 | 0.619 | 15 (35) | 28 (62) | 3244-2_K_03161 |
| sfaG | S/F1C fimbrial adhesin minor pilin SfaG/FocF | 0.0118 | 0.619 | 15 (35) | 28 (62) | 3244-2_K_03159 |
| sfaD | S/F1C fimbrial minor subunit | 0.0118 | 0.619 | 15 (35) | 28 (62) | 3244-2_K_03162 |
| ydiV | Putative anti-FlhC(2)FlhD(4) factor YdiV | 0.0118 | 0.619 | 15 (35) | 28 (62) | 3244-2_K_03156 |
| hifA | fimbrial protein | 0.0095 | 0.619 | 11 (26) | 24 (53) | 3244-2_K_02738 |
| Other virulence-related genes | ||||||
| shlB | Hemolysin transport | 0.0100 | 0.619 | 16 (37) | 30 (67) | 3230-3_K_03314 |
| cbeA | Toxin/antitoxin system | 0.0128 | 0.619 | 33 (77) | 43 (96) | 3244-2_K_03419 |
| yeeW | Toxin/antitoxin protein | 0.0106 | 0.619 | 28 (65) | 40 (89) | 3244-2_K_03421 |
| yeeJ | Adhesin | 0.0079 | 0.619 | 29 (67) | 41 (91) | 3244-2_K_00508 |
| wgrG | Type VI secretion system protein | 0.0088 | 0.619 | 4 (9) | 15 (33) | 3244-2_K_04080 |
| klcA | Anti-restriction protein | 0.0068 | 0.619 | 34 (79) | 44 (98) | 3014-3_K_04082 |
| cbtA | Toxin/antitoxin system | 0.0064 | 0.619 | 32 (74) | 43 (96) | 3244-2_K_04704 |
| LPS-biosynthesis | ||||||
| gspA_3 | Secretion pathway protein | 0.0015 | 0.619 | 30 (70) | 43 (96) | 3244-2_K_02772 |
| gspA_4 | Secretion pathway protein | 0.0015 | 0.619 | 30 (70) | 43 (96) | 3244-2_K_00614 |
| rfaY | Lps biosynthesis protein | 0.0015 | 0.619 | 30 (70) | 43 (96) | 3244-2_K_02773 |
| epsJ | Glycosyltransferase | 0.0028 | 0.619 | 26 (60) | 40 (89) | 3244-2_K_02775 |
| gspA_2 | Secretion pathway protein | 0.0028 | 0.619 | 26 (60) | 40 (89) | 3244-2_K_02774 |
| rfaL | Lps biosynthesis protein | 0.0028 | 0.619 | 26 (60) | 40 (89) | 3244-2_K_02776 |
| Phage-related genes | ||||||
| - | Bacteriophage Replication protein O | 0.0056 | 0.619 | 27 (63) | 40 (89) | 3014-3_K_04219 |
| - | Phage replication protein | 0.0057 | 0.619 | 23 (53) | 37 (82) | 3014-3_K_04220 |
| alpA | Prophage regulatory protein | 0.0007 | 0.619 | 20 (47) | 37 (82) | 3244-2_K_03100 |
| Metabolism-related genes | ||||||
| bglA | Metabolism protein | 0.0079 | 0.619 | 29 (67) | 41 (91) | 3244-2_K_00072 |
| bglH | Metabolism protein | 0.0079 | 0.619 | 29 (67) | 41 (91) | 3244-2_K_00073 |
| yidP | Transcriptional regulator | 0.0079 | 0.619 | 29 (67) | 41 (91) | 3244-2_K_00077 |
| yfjR | Transcriptional regulator | 0.0082 | 0.619 | 20 (46) | 34 (76) | 3244-2_K_03712 |
| ykfF | Hypothetical protein | 0.0089 | 0.619 | 19 (44) | 33 (73) | 3244-2_K_03710 |
| Hypothetical protein | 0.0017 | 0.619 | 20 (47) | 36 (80) | 3244-2_K_03711 | |
| rspA | Starvation sensing protein | 0.0038 | 0.619 | 25 (58) | 39 (87) | 3244-2_K_01532 |
| yjjL | Inner membrane transport protein | 0.0038 | 0.619 | 25 (58) | 39 (87) | 3244-2_K_01531 |
| - | Putative inner membrane protein | 0.0024 | 0.619 | 23 (53) | 38 (84) | 3244-2_K_03387 |
| - | Cytoplasmic protein | 0.0047 | 0.619 | 24 (56) | 38 (84) | 3244-2_K_03386 |
| rbsK | Ribokinase | 0.0047 | 0.619 | 24 (56) | 38 (84) | 3244-2_K_03385 |
| Replication | ||||||
| crfC | Replication protein | 0.0122 | 0.619 | 23 (53) | 36 (80) | 3244-2_K_03407 |
| Chemotaxis protein | 0.0057 | 0.619 | 23 (53) | 37 (82) | 3244-2_K_03408 | |
| Other and unknown function | ||||||
| tnpB | Transposase | 0.0098 | 0.619 | 26 (60) | 14 (31) | 3805-3_K_04559 |
| ompD | Porin protein | 0.0057 | 0.619 | 23 (53) | 37 (82) | 3244_2_K_00430 |
| - | Hypothetical protein | 0.0118 | 0.619 | 15 (35) | 28 (62) | 3244-2_K_03094 |
| - | Hypothetical protein | 0.0137 | 0.619 | 22 (51) | 35 (78) | 3244-2_K_03400 |
| higA | Toxin/antitoxin system | 0.0122 | 0.619 | 23 (53) | 36 (80) | 3244-2_K_00724 |
| Overrepresented in non-rUTI isolates | ||||||
| pipB | Type III secretion system protein | 0.0106 | 0.619 | 15 (35) | 5 (11) | 3212-3_K_02120 |
| - | Hypothetical protein (prophage) | 0.0112 | 0.619 | 6 (14) | 0 (0) | MN187550_Orf34 |
| waaZ | Lps biosynthesis | 0.0112 | 0.619 | 6 (14) | 0 (0) | CP046003_GJD94_01225 |
| waaY | Lps biosynthesis | 0.0112 | 0.619 | 6 (14) | 0 (0) | CP046003_GJD94_01220 |
| waaJ | Lps biosynthesis | 0.0112 | 0.619 | 6 (14) | 0 (0) | CP046003_GJD94_01215 |
| waaO | Lps biosynthesis | 0.0112 | 0.619 | 6 (14) | 0 (0) | CP046003_GJD94_01210 |
| yibB | RfaH-regulated high-temperature protein | 0.0032 | 0.619 | 10 | 1 | CP046003_GJD94_01255 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nielsen, K.L.; Stegger, M.; Kiil, K.; Lilje, B.; Ejrnæs, K.; Leihof, R.F.; Skjøt-Rasmussen, L.; Godfrey, P.; Monsen, T.; Ferry, S.; et al. Escherichia coli Causing Recurrent Urinary Tract Infections: Comparison to Non-Recurrent Isolates and Genomic Adaptation in Recurrent Infections. Microorganisms 2021, 9, 1416. https://doi.org/10.3390/microorganisms9071416
Nielsen KL, Stegger M, Kiil K, Lilje B, Ejrnæs K, Leihof RF, Skjøt-Rasmussen L, Godfrey P, Monsen T, Ferry S, et al. Escherichia coli Causing Recurrent Urinary Tract Infections: Comparison to Non-Recurrent Isolates and Genomic Adaptation in Recurrent Infections. Microorganisms. 2021; 9(7):1416. https://doi.org/10.3390/microorganisms9071416
Chicago/Turabian StyleNielsen, Karen Leth, Marc Stegger, Kristoffer Kiil, Berit Lilje, Karen Ejrnæs, Rikke Fleron Leihof, Line Skjøt-Rasmussen, Paul Godfrey, Tor Monsen, Sven Ferry, and et al. 2021. "Escherichia coli Causing Recurrent Urinary Tract Infections: Comparison to Non-Recurrent Isolates and Genomic Adaptation in Recurrent Infections" Microorganisms 9, no. 7: 1416. https://doi.org/10.3390/microorganisms9071416