
Elucidating the Role of Transcriptomic Networks and DNA Methylation in Collagen Deposition of Dezhou Donkey Skin

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Animals and Sample Collection
2.3. Sequencing and Analysis of Methylomes
2.4. Methylation Data Analysis
2.5. Sequencing and Processing of Transcriptome Data
2.6. Correlation of DMRs and DEGs between Groups
2.7. Bisulfite Sequencing PCR (BSP)
2.8. Statistical Analysis
3. Results
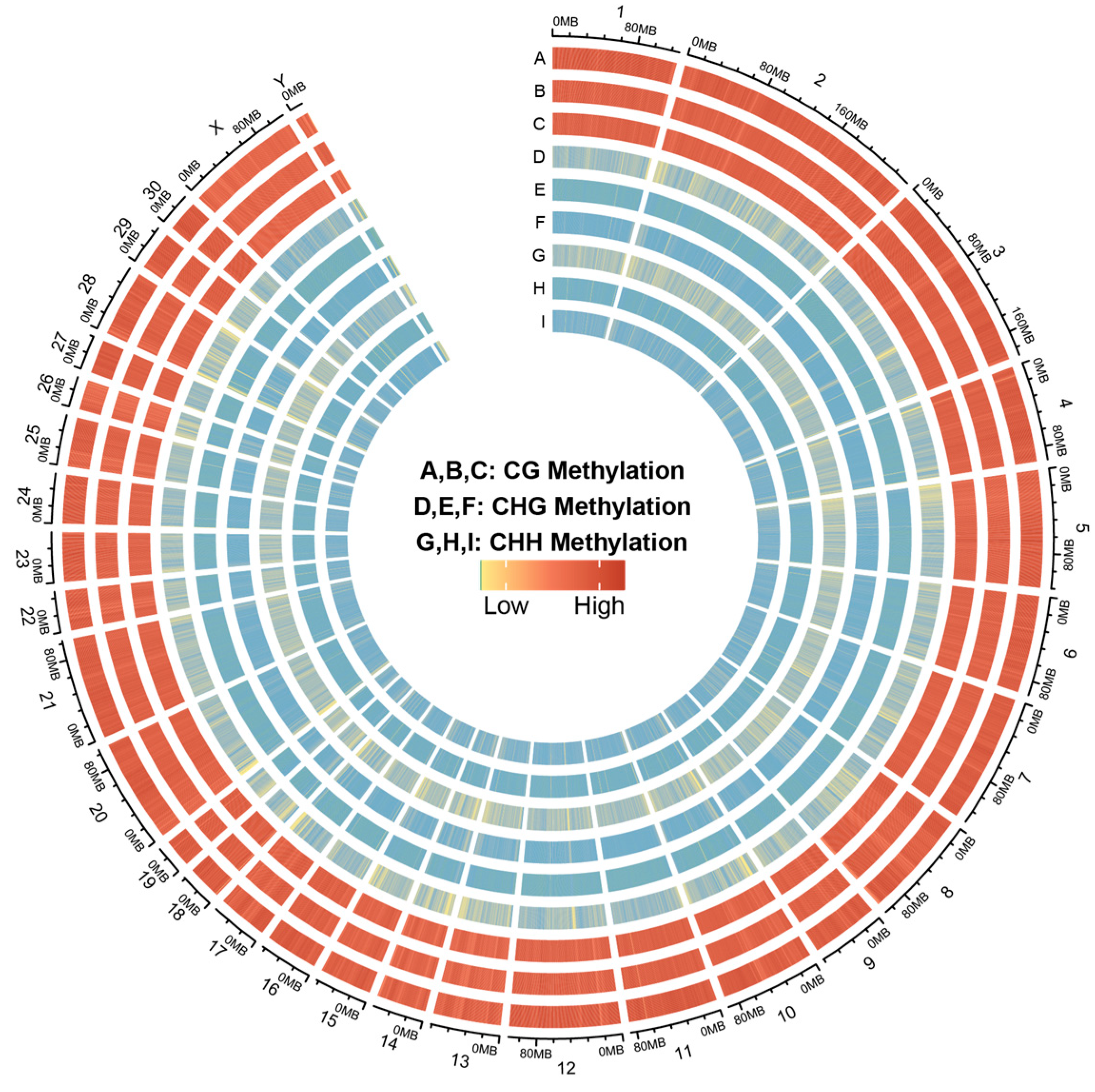
3.1. The DNA Methylation Atlas of Skin Tissues of Dezhou Donkey at Different Developmental Periods
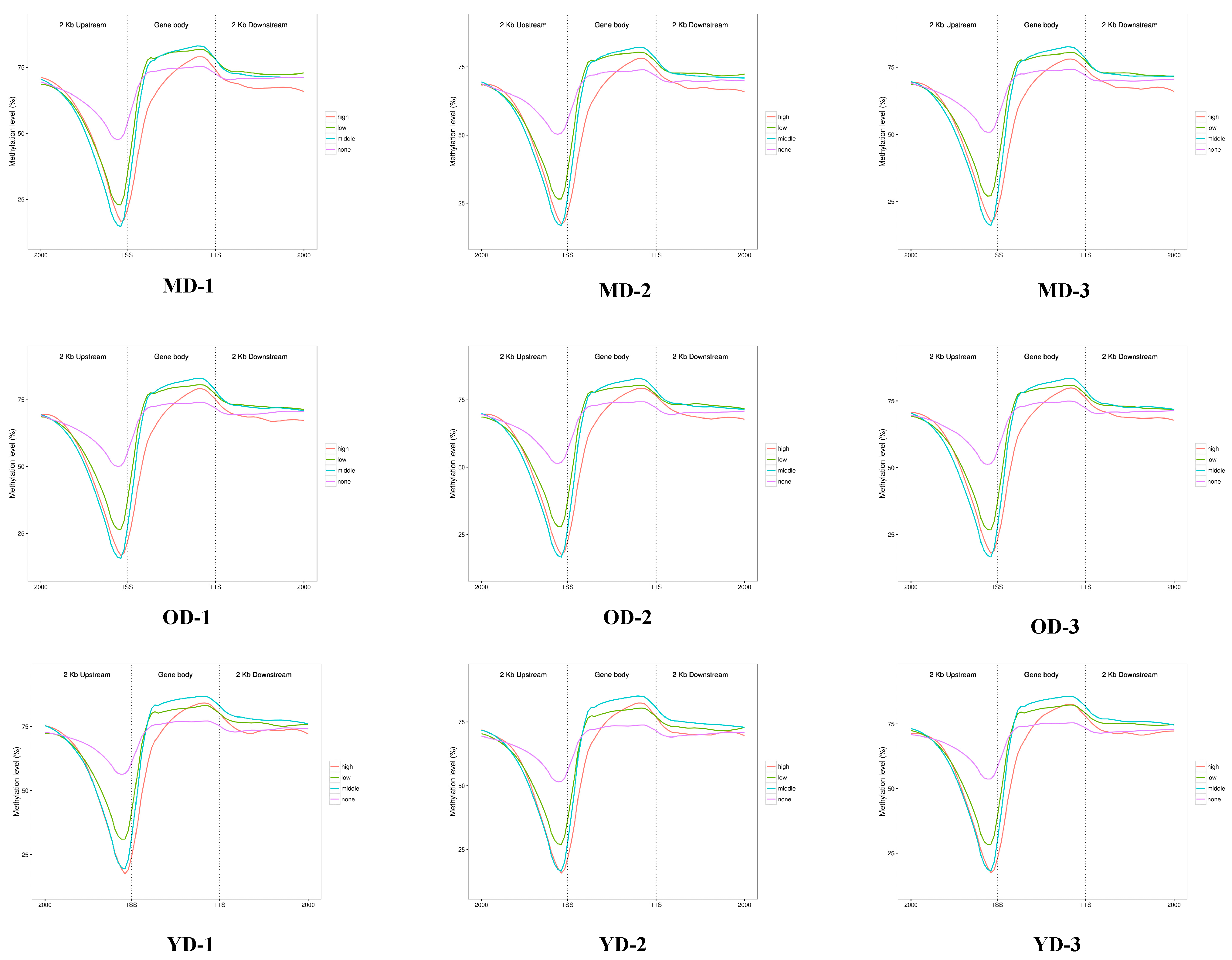
3.2. Global DNA Methylation Patterns of Skin Tissues in Dezhou Donkey
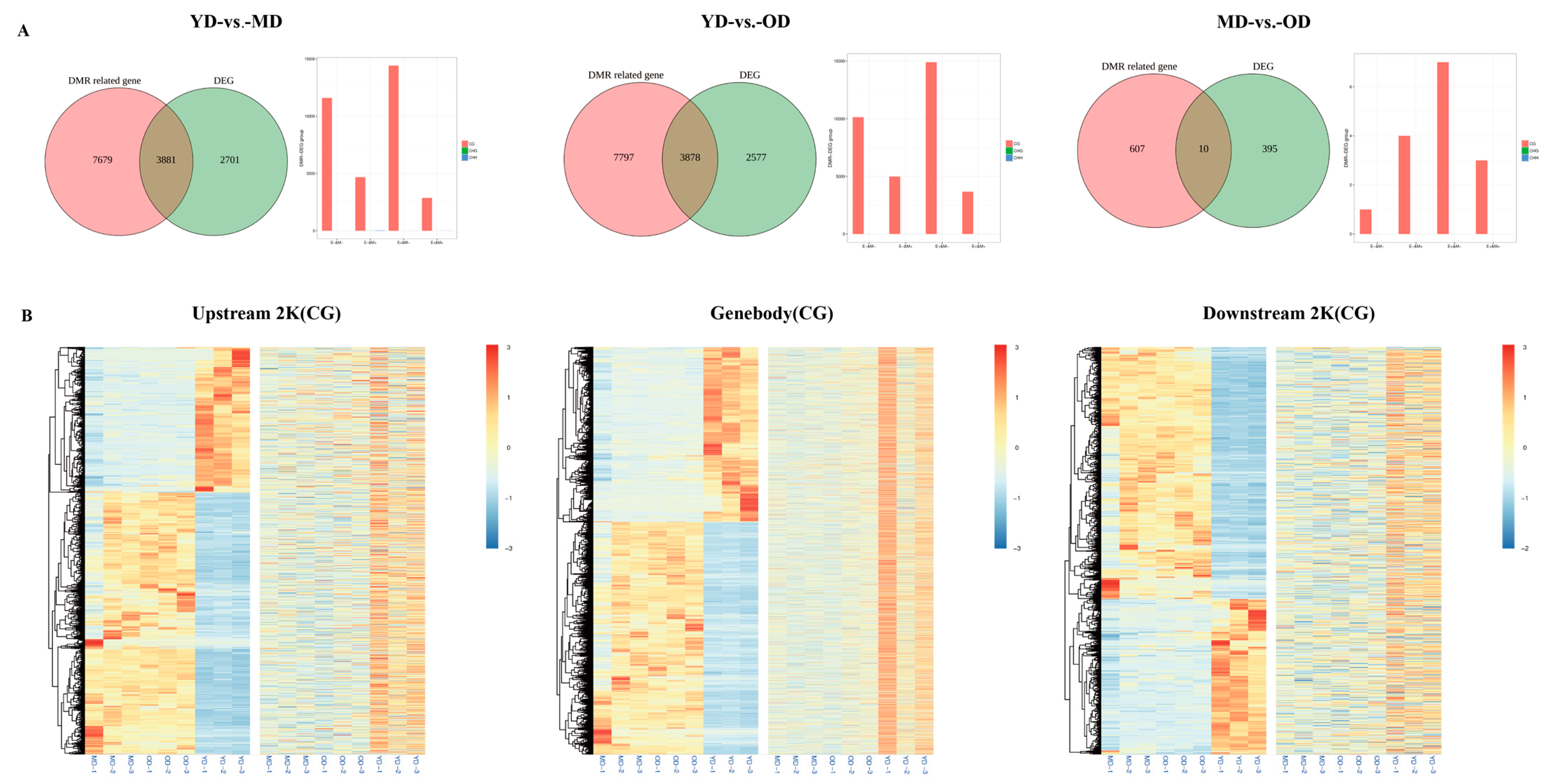
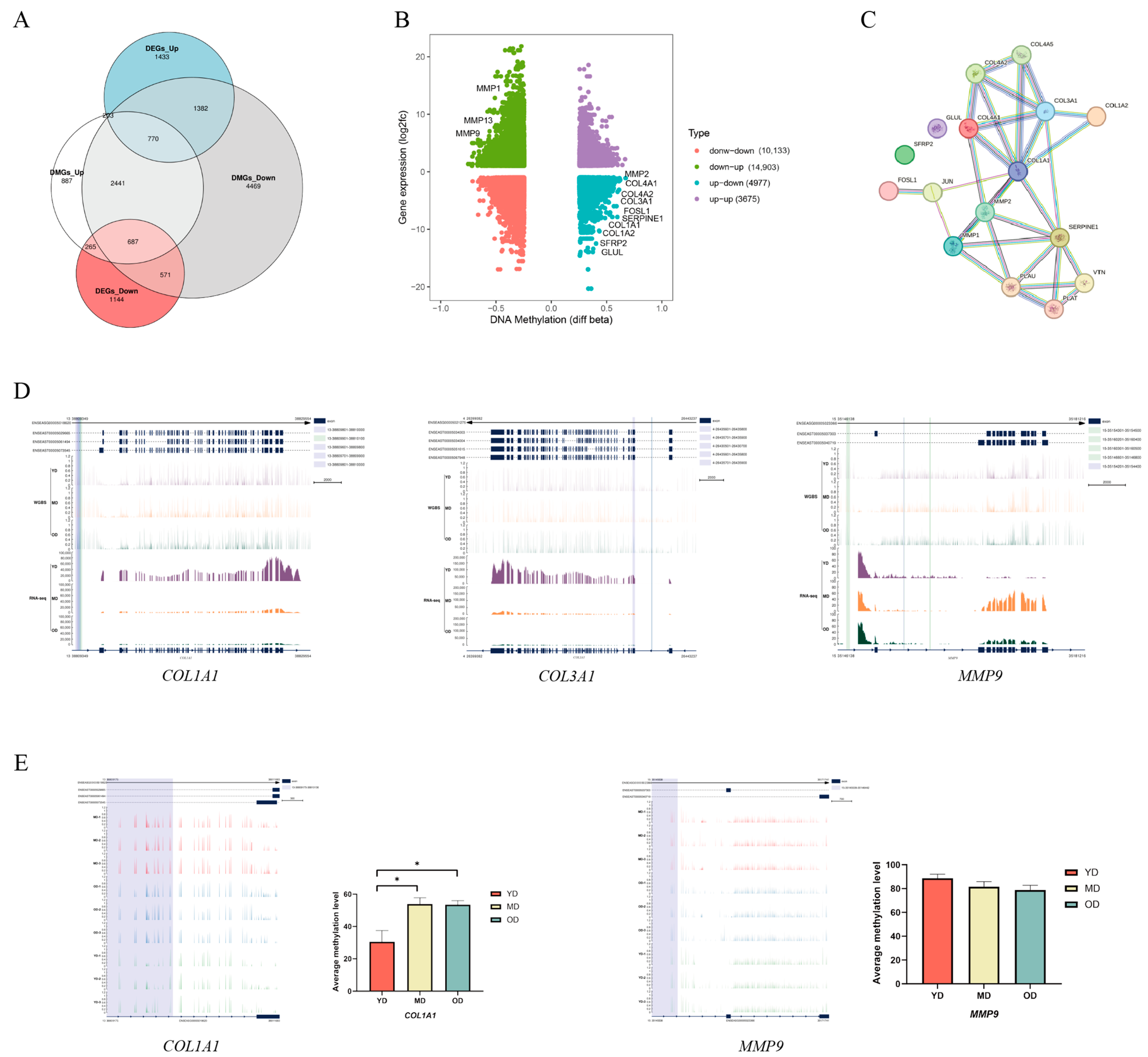
3.3. WGBS-Transcriptomic Data Integration
3.4. Analysis of the Relationship between DEGs and DNA Methylation in Skin Tissues at Different Developmental Stages
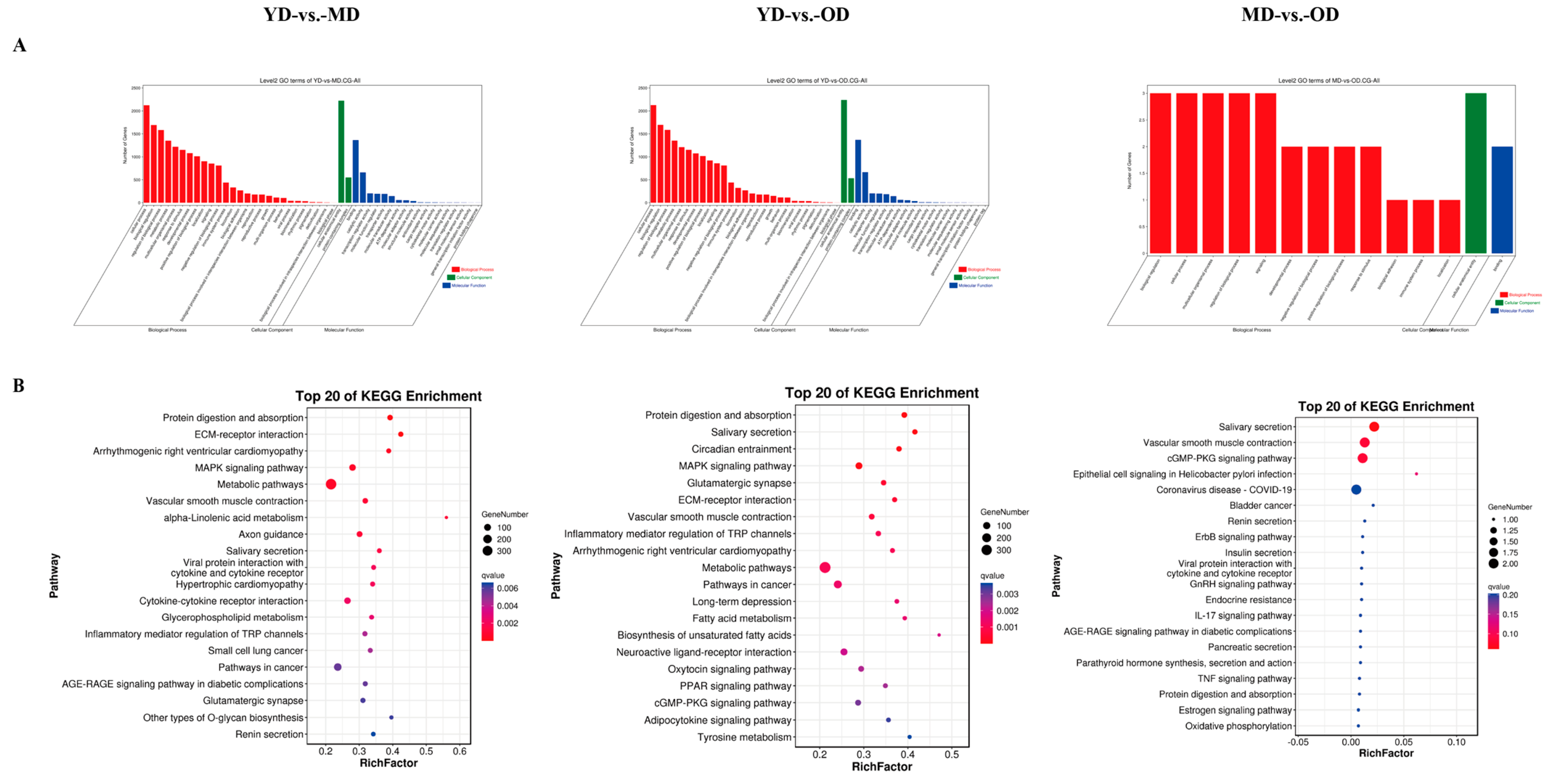
3.5. Enrichment Analysis of Methylation-Related DEGs
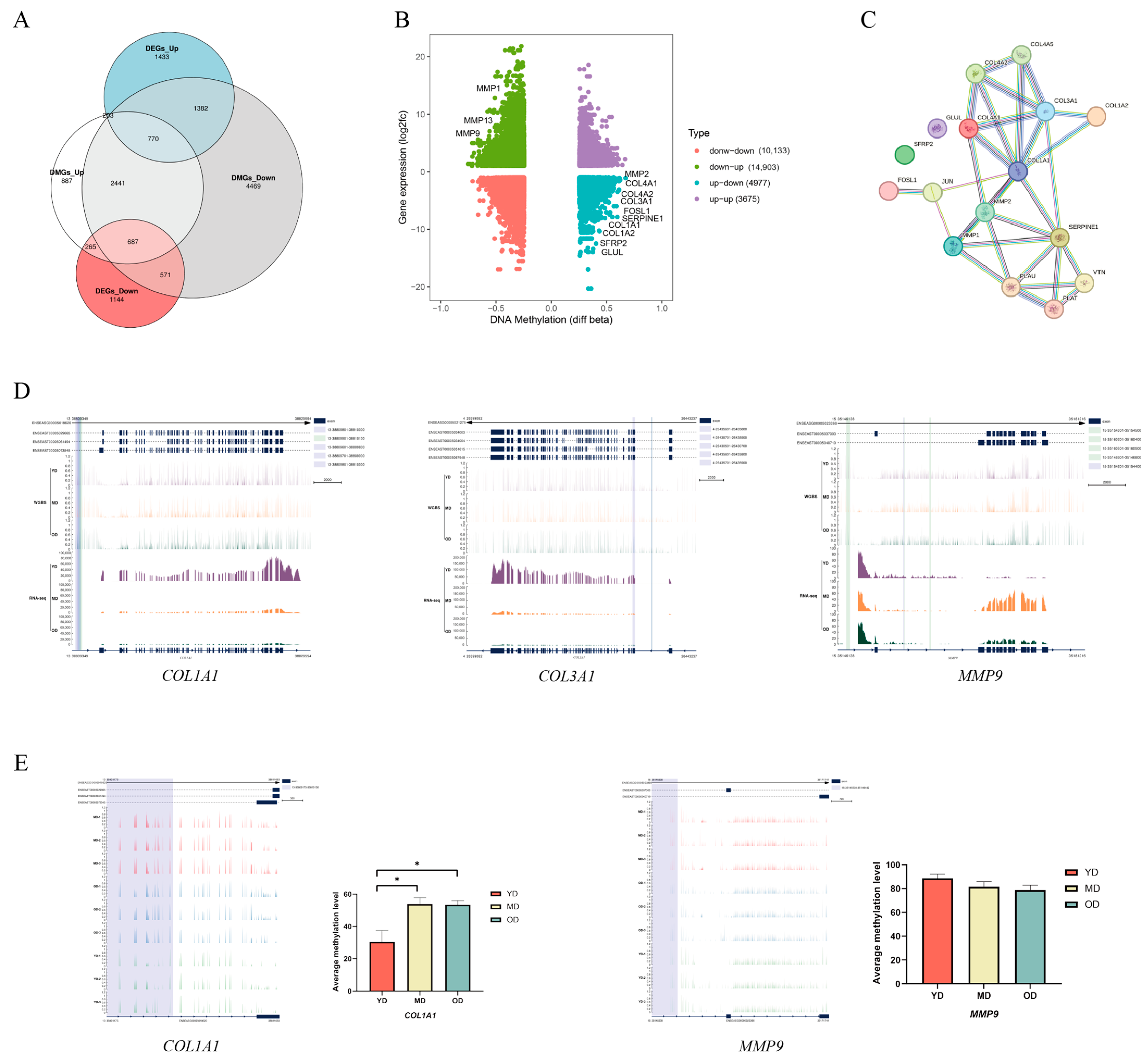
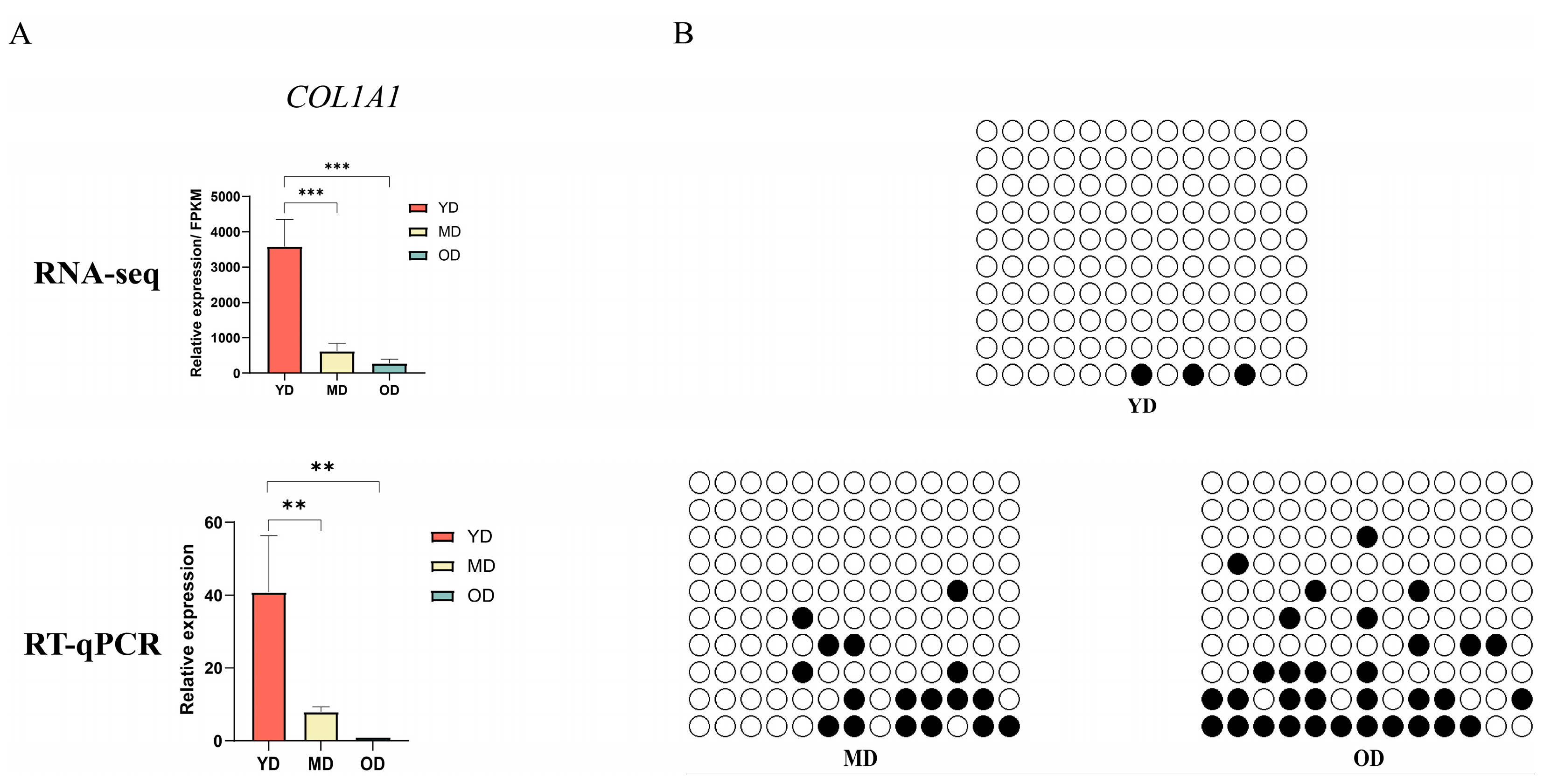
3.6. Candidate DMGs Associated with Collagen in Dezhou Donkey Skin
3.7. DNA Methylation of COL1A1 Promoter Region
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seyiti, S.; Kelimu, A. Donkey Industry in China: Current Aspects, Suggestions and Future Challenges. J. Equine Vet. Sci. 2021, 102, 103642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cui, S.; Cheng, X.-L.; Wei, F.; Ma, S. An Optimized TaqMan Real-Time PCR Method for Authentication of ASINI CORII COLLA (Donkey-Hide Gelatin). J. Pharm. Biomed. Anal. 2019, 170, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, F.; Gong, L.; Hang, B.; Li, D.; Chi, L. Species-Specific Identification of Collagen Components in Colla Corii Asini Using a Nano-Liquid Chromatography Tandem Mass Spectrometry Proteomics Approach. Int. J. Nanomed. 2017, 12, 4443–4454. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Song, J.; Liang, M.; Ma, F.; Mao, X.; Ma, C.W.; Zhang, W.; Huang, Z. Overview of Beverages with Anti-Aging Functions in Chinese Market. Rejuvenation Res. 2014, 17, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liang, H.; Khan, M.Z.; Ren, W.; Huang, B.; Chen, Y.; Xing, S.; Zhan, Y.; Wang, C. Comprehensive transcriptomic analysis unveils the interplay of mRNA and LncRNA expression in shaping collagen organization and skin development in Dezhou donkeys. Front. Genet. 2024, 15, 1335591. [Google Scholar]
- Huang, B.; Khan, M.Z.; Chai, W.; Ullah, Q.; Wang, C. Exploring Genetic Markers: Mitochondrial DNA and Genomic Screening for Biodiversity and Production Traits in Donkeys. Animals. 2023, 13, 2725. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.A.; Roberts, M.S. The structure and function of skin. In Dermatological and Transdermal Formulations; CRC Press: Boca Raton, FL, USA, 2002; pp. 19–58. [Google Scholar]
- Mayne, R.; Burgeson, R.E. (Eds.) Structure and Function of Collagen Types; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Yang, Y.-L.; Rong, Z.; Kui, L. Future Livestock Breeding: Precision Breeding Based on Multi-Omics Information and Population Personalization. J. Integr. Agric. 2017, 16, 2784–2791. [Google Scholar] [CrossRef]
- Lockhart, D.J.; Winzeler, E.A. Genomics, Gene Expression and DNA Arrays. Nature 2000, 405, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Huang, Y.; Yang, Z.; Ma, Y.; Chaogetu, B.; Zhuoma, Z.; Chen, H. RNA-Seq Analysis Identifies Differentially Expressed Genes in Subcutaneous Adipose Tissue in Qaidaford Cattle, Cattle-Yak, and Angus Cattle. Animals 2019, 9, 1077. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.; Cánovas, A.; Rincón, G.; Medrano, J.F. RNA-Sequencing: A Tool to Explore New Frontiers in Animal Genetics. Livest. Sci. 2014, 166, 206–216. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA Sequencing: The Teenage Years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Qu, H.; Ma, Q.; Zhu, M.; Li, M.; Zhan, Y.; Liu, Z.; Xu, J.; Yao, H.; Li, Z.; et al. RNA-Seq Analysis Identifies Differentially Expressed Gene in Different Types of Donkey Skeletal Muscles. Anim. Biotechnol. 2023, 34, 1786–1795. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, H.; Zhang, X.; Yang, L.; Liu, Y.; Liu, S.; Sun, Y.; Zhao, C. An Analysis of Skin Thickness in the Dezhou Donkey Population and Identification of Candidate Genes by RNA-Seq. Anim. Genet. 2022, 53, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Wang, J.; Li, Y.; Yang, C.; Zhang, R.; Wang, X.; Ju, Z.; Jiang, Q.; Huang, J.; Wang, C.; et al. Integrated Analysis of mRNA and miRNA in Testis and Cauda Epididymidis Reveals Candidate Molecular Markers Associated with Reproduction in Dezhou Donkey. Livest. Sci. 2020, 234, 103885. [Google Scholar] [CrossRef]
- Yu, J.; Yang, G.; Li, S.; Li, M.; Ji, C.; Liu, G.; Wang, Y.; Chen, N.; Lei, C.; Dang, R. Identification of Dezhou Donkey Muscle Development-Related Genes and Long Non-Coding RNA Based on Differential Expression Analysis. Anim. Biotechnol. 2023, 34, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, H.; Guo, Y.; Huang, J.; Sun, Y.; Min, J.; Wang, J.; Fang, X.; Zhao, Z.; Wang, S.; et al. Donkey Genomes Provide New Insights into Domestication and Selection for Coat Color. Nat. Commun. 2020, 11, 6014. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, T.; Sparkes, R.; Shapiro, L. Reactivation of an Inactive Human X Chromosome: Evidence for X Inactivation by DNA Methylation. Science 1981, 211, 393–396. [Google Scholar] [CrossRef]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA Methylation and DNA Methyltransferases. Epigenetics Chromatin 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-Mediated Active DNA Demethylation: Mechanism, Function and Beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Yang, S.; Huang, Y.; He, H.; Lei, C.; Cheng, H. Research and Application of DNA Methylation in Animal Genetics and Breeding. China Cattle Sci. 2016, 42, 51–54. [Google Scholar]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.; Jung, E.J.; Mattei, A.L.; Bailly, N.; Liao, J.; Martin, E.J.; Giesselmann, P.; Brändl, B.; Stamenova, E.K.; Müller, F.-J.; et al. TETs compete with DNMT3 activity in pluripotent cells at thousands of methylated somatic enhancers. Nat. Genet. 2020, 52, 819–827. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, T.; Wu, H.; Zhang, J.; Zhou, C.; Jiang, A.; Li, R.; Li, X. Genome-Wide DNA Methylation Changes between the Superficial and Deep Backfat Tissues of the Pig. Int. J. Mol. Sci. 2012, 13, 7098–7108. [Google Scholar] [CrossRef]
- Zhang, M.; Li, D.; Zhai, Y.; Wang, Z.; Ma, X.; Zhang, D.; Li, G.; Han, R.; Jiang, R.; Li, Z.; et al. The Landscape of DNA Methylation Associated with the Transcriptomic Network of Intramuscular Adipocytes Generates Insight into Intramuscular Fat Deposition in Chicken. Front. Cell Dev. Biol. 2020, 8, 206. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Zhu, C.; Li, F.; Liu, Z.; Li, X.; Shen, X.; Wu, Z.; Fu, M.; Xu, D.; et al. DNA Demethylation of Myogenic Genes May Contribute to Embryonic Leg Muscle Development Differences between Wuzong and Shitou Geese. Int. J. Mol. Sci. 2023, 24, 7188. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; E Garrett-Bakelman, F.; E Figueroa, M.; Melnick, A.; E Mason, C. MethylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Adams, S.L. Collagen Gene Expression. Am. J. Respir. Cell Mol. Biol. 1989, 1, 161–168. [Google Scholar] [CrossRef]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Smith, Z.D.; Chan, M.M.; Humm, K.C.; Karnik, R.; Mekhoubad, S.; Regev, A.; Eggan, K.; Meissner, A. DNA methylation dynamics of the human preimplantation embryo. Nature 2014, 511, 611–615. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef]
- Zhang, X.; Nie, Y.; Cai, S.; Ding, S.; Fu, B.; Wei, H.; Chen, L.; Liu, X.; Liu, M.; Yuan, R.; et al. Earlier Demethylation of Myogenic Genes Contributes to Embryonic Precocious Terminal Differentiation of Myoblasts in Miniature Pigs. FASEB J. 2019, 33, 9638–9655. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Halušková, J.; Holečková, B.; Staničová, J. DNA Methylation Studies in Cattle. J. Appl. Genet. 2021, 62, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Zhang, Z.; Zou, Z.; Lv, C.; Dong, Q.; He, Q.; Yi, Y.; Li, Y.; Wang, H.; Zhu, B. Kinetics and Mechanisms of Mitotic Inheritance of DNA Methylation and Their Roles in Aging-Associated Methylome Deterioration. Cell Res. 2020, 30, 980–996. [Google Scholar] [CrossRef]
- Liu, J.; Luo, C.; Yin, Z.; Li, P.; Wang, S.; Chen, J.; He, Q.; Zhou, J. Downregulation of Let 7b Promotes COL1A1 and COL1A2 Expression in Dermis and Skin Fibroblasts during Heat Wound Repair. Mol. Med. Rep. 2016, 13, 2683–2688. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Tromp, G. Type III Collagen (COL3A1): Gene and Protein Structure, Tissue Distribution, and Associated Diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Abreu-Velez, A.M.; Howard, M.S. Collagen IV in Normal Skin and in Pathological Processes. N. Am. J. Med. Sci. 2012, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayyat, W.; Pirkkanen, J.; Dougherty, J.; Dickinson, N.; Khaper, N.; Lees, S.J.; Mendonca, M.S.; Boreham, D.R.; Chun Tai, T.; Thome, C.; et al. Overexpression of FRA1 (FOSL1) Leads to Global Transcriptional Perturbations, Reduced Cellular Adhesion, and Altered Cell Cycle Progression. Cells 2023, 12, 2344. [Google Scholar] [CrossRef] [PubMed]
- Freeberg, M.A.T.; Farhat, Y.M.; Easa, A.; Kallenbach, J.G.; Malcolm, D.W.; Buckley, M.R.; Benoit, D.S.W.; Awad, H.A. Serpine1 Knockdown Enhances MMP Activity after Flexor Tendon Injury in Mice: Implications for Adhesions Therapy. Sci. Rep. 2018, 8, 5810. [Google Scholar] [CrossRef]
- Lauer Fields, J.L.; Juska, D.; Fields, G.B. Matrix Metalloproteinases and Collagen Catabolism. Biopolymers 2002, 66, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Fréchet, M.; Warrick, E.; Vioux, C.; Chevallier, O.; Spatz, A.; Benhamou, S.; Sarasin, A.; Bernerd, F.; Magnaldo, T. Overexpression of Matrix Metalloproteinase 1 in Dermal Fibroblasts from DNA Repair-Deficient/Cancer-Prone Xeroderma Pigmentosum Group C Patients. Oncogene 2008, 27, 5223–5232. [Google Scholar] [CrossRef]
- Han, H.; Du, B.; Pan, X.; Liu, J.; Zhao, Q.; Lian, X.; Qian, M.; Liu, M. CADPE Inhibits PMA-Stimulated Gastric Carcinoma Cell Invasion and Matrix Metalloproteinase-9 Expression by FAK/MEK/ERK–Mediated AP-1 Activation. Mol. Cancer Res. 2010, 8, 1477–1488. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) | Production |
|---|---|---|
| COL1A1-CpG1-F | TTTTATTAAGATGGTATAAAAGGGG | 163 bp |
| COL1A1-CpG1-R | ATATCTAAACCCTAAACATATAAACTCTT |
| Sample | Raw Data (bp) | Clean Data | Clean Data (%) | Mapped Ratio (%) | Q20 (%) |
|---|---|---|---|---|---|
| YD-1 | 72,478,223,100 | 70,905,443,678 | 97.83% | 90.49 | 96.9 |
| YD-2 | 83,599,930,500 | 81,884,673,906 | 97.95% | 89.94 | 96.49 |
| YD-3 | 81,479,674,800 | 79,816,131,823 | 97.96% | 90.41 | 96.87 |
| MD-1 | 75,431,070,600 | 73,914,308,509 | 97.99% | 90.29 | 96.7 |
| MD-2 | 80,349,082,500 | 78,746,354,773 | 98.01% | 90.22 | 96.64 |
| MD-3 | 77,751,545,400 | 75,957,186,447 | 97.69% | 90.12 | 96.25 |
| OD-1 | 79,657,478,400 | 77,820,858,668 | 97.69% | 89.57 | 95.91 |
| OD-2 | 75,182,412,600 | 73,607,402,411 | 97.91% | 89.69 | 96.07 |
| OD-3 | 72,051,872,400 | 69,294,945,450 | 96.17% | 89.24 | 96.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Ren, W.; Peng, Y.; Khan, M.Z.; Liang, H.; Zhang, Y.; Liu, X.; Chen, Y.; Kou, X.; Wang, L.; et al. Elucidating the Role of Transcriptomic Networks and DNA Methylation in Collagen Deposition of Dezhou Donkey Skin. Animals 2024, 14, 1222. https://doi.org/10.3390/ani14081222
Wang X, Ren W, Peng Y, Khan MZ, Liang H, Zhang Y, Liu X, Chen Y, Kou X, Wang L, et al. Elucidating the Role of Transcriptomic Networks and DNA Methylation in Collagen Deposition of Dezhou Donkey Skin. Animals. 2024; 14(8):1222. https://doi.org/10.3390/ani14081222
Chicago/Turabian StyleWang, Xinrui, Wei Ren, Yongdong Peng, Muhammad Zahoor Khan, Huili Liang, Yigang Zhang, Xiaotong Liu, Yinghui Chen, Xiyan Kou, Liyuan Wang, and et al. 2024. "Elucidating the Role of Transcriptomic Networks and DNA Methylation in Collagen Deposition of Dezhou Donkey Skin" Animals 14, no. 8: 1222. https://doi.org/10.3390/ani14081222