

Potential Inhibitors of Lumpy Skin Disease’s Viral Protein (DNA Polymerase): A Combination of Bioinformatics Approaches

 ,
,  , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Target Selection and Validation
2.2. Ligand Selection
2.3. Ligand Preparation
2.4. Protein Preparation
2.5. Site Map Analysis
2.6. Receptor Grid Generation
2.7. Molecular Docking
2.7.1. Virtual Screening and Ligand Docking
2.7.2. Free Energy Calculation by MM-GBSA
2.7.3. ADMET Profiling of Novel Antiviral Compounds against LSDV
2.7.4. Molecular Dynamics (MDs) Simulation
3. Results
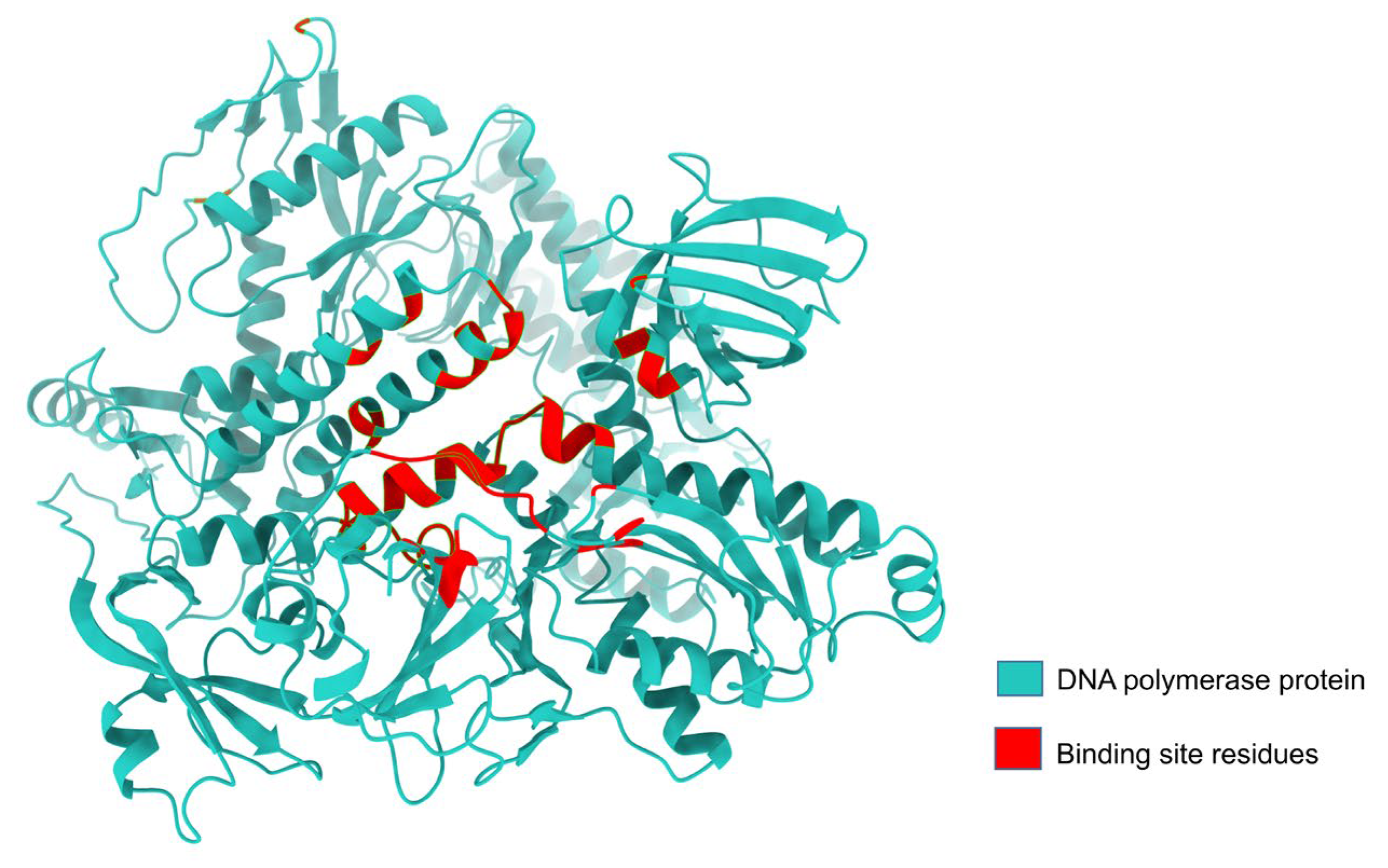
3.1. LSDV DNA Polymerase Protein Has a Groove-like Active Site
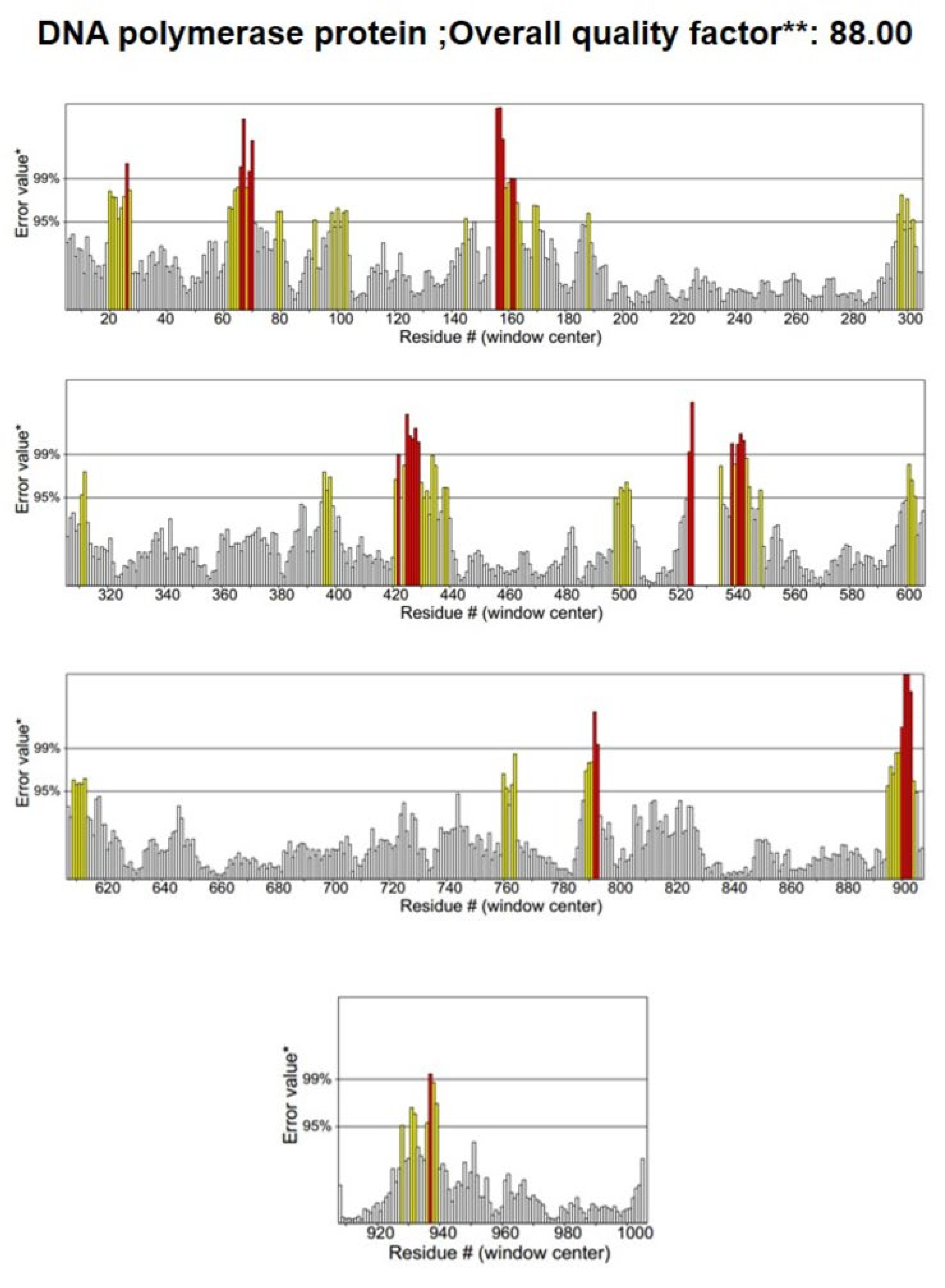
3.2. Structure Validation
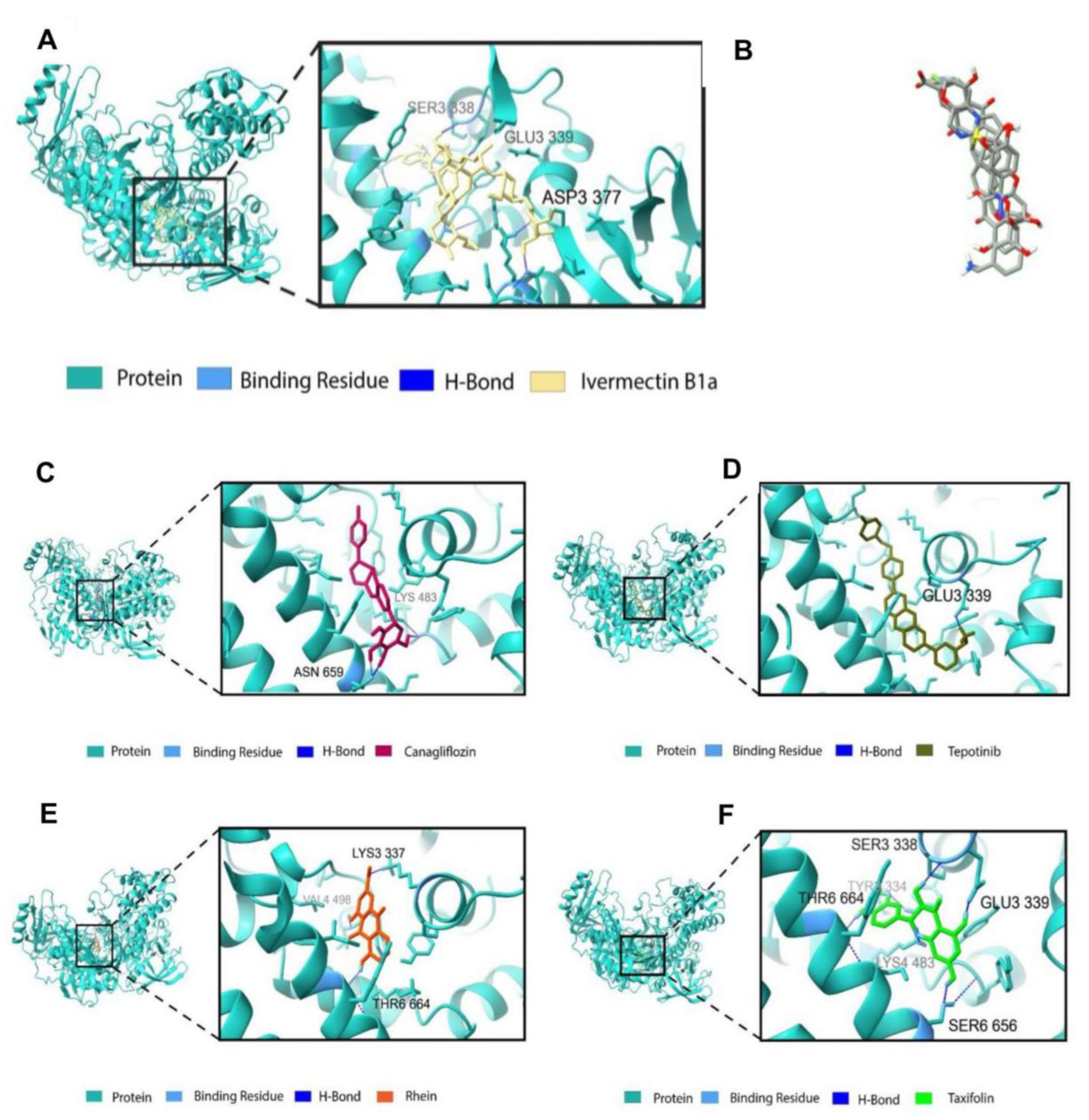
3.3. Binding Profile Analysis of Bonded Interactions
3.4. Binding Free Energy Calculations for Non-Bonded Interactions
3.5. ADMET Profiling of Novel Antiviral Compounds against LSDV
3.6. Molecular Dynamics (MDs) Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Das, M.; Chowdhury, M.S.R.; Akter, S.; Mondal, A.K.; Uddin, M.J.; Rahman, M.M.; Rahman, M.M. An updated review on lumpy skin disease: Perspective of Southeast Asian countries. J. Adv. Biotechnol. Exp. Ther. 2021, 4, 322–333. [Google Scholar] [CrossRef]
- Molini, U.; Boshoff, E.; Niel, A.P.; Phillips, J.; Khaiseb, S.; Settypalli, T.B.; Dundon, W.G.; Cattoli, G.; Lamien, C.E. Detection of lumpy skin disease virus in an asymptomatic eland (Taurotragus oryx) in Namibia. J. Wildl. Dis. 2021, 57, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.S.; Oh, Y.; Lee, T.G.; Bae, D.Y.; Tark, D.; Cho, H.S. Serological and molecular prevalence of lumpy skin disease virus in Korean water deer, native and dairy cattle in Korea. Korean J. Vet. Serv. 2022, 45, 133–137. [Google Scholar] [CrossRef]
- Dao, T.D.; Tran, L.H.; Nguyen, H.D.; Hoang, T.T.; Nguyen, G.H.; Tran KV, D.; Nguyen, H.X.; Van Dong, H.; Bui, A.N.; Bui, V.N. Characterization of Lumpy skin disease virus isolated from a giraffe in Vietnam. Transbound. Emerg. Dis. 2022, 69, e3268–e3272. [Google Scholar] [CrossRef]
- Sprygin, A.; Pestova, Y.; Wallace, D.; Tuppurainen, E.; Kononov, A. Transmission of lumpy skin disease virus: A short review. Virus Res. 2019, 269, 197637. [Google Scholar] [CrossRef] [PubMed]
- Hasib, F.M.; Islam, M.S.; Das, T.; Rana, E.A.; Uddin, M.H.; Bayzid, M.; Nath, C.; Hossain, M.A.; Masuduzzaman, M.; Das, S.; et al. Lumpy skin disease outbreak in cattle population of Chattogram, Bangladesh. Vet. Med. Sci. 2021, 7, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Venter, E.H.; Coetzer, J.A.; Bell-Sakyi, L. Lumpy skin disease: Attempted propagation in tick cell lines and presence of viral DNA in field ticks collected from naturally-infected cattle. Ticks Tick-Borne Dis. 2015, 6, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Bernardo, B.; Haga, I.R.; Wijesiriwardana, N.; Basu, S.; Larner, W.; Diaz, A.V.; Langlands, Z.; Denison, E.; Stoner, J.; White, M. Quantifying and modeling the acquisition and retention of lumpy skin disease virus by hematophagus insects reveals clinically but not subclinically affected cattle are promoters of viral transmission and key targets for control of disease outbreaks. J. Virol. 2021, 95, e02185-18. [Google Scholar]
- Datten, B.; Chaudhary, A.A.; Sharma, S.; Singh, L.; Rawat, K.D.; Ashraf, M.S.; Alneghery, L.M.; Aladwani, M.O.; Rudayni, H.A.; Dayal, D.; et al. An Extensive Examination of the Warning Signs, Symptoms, Diagnosis, Available Therapies, and Prognosis for Lumpy Skin Disease. Viruses 2023, 15, 604. [Google Scholar] [CrossRef]
- Wilhelm, L.; Ward, M.P. The Spread of Lumpy Skin Disease Virus across Southeast Asia: Insights from Surveillance. Transbound. Emerg. Dis. 2023, 2023, 3972359. [Google Scholar] [CrossRef]
- Chouhan, C.S.; Parvin, M.S.; Ali, M.Y.; Sadekuzzaman, M.; Chowdhury, M.G.A.; Ehsan, M.A.; Islam, M.T. Epidemiology and economic impact of lumpy skin disease of cattle in Mymensingh and Gaibandha districts of Bangladesh. Transbound. Emerg. Dis. 2022, 69, 3405–3418. [Google Scholar] [CrossRef] [PubMed]
- Tadesse Degu, B.M.; Fesseha, H. Epidemiological status and economic impact of lumpy skin disease-review. Int. J. Recent Biotechnol. 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Liang, Z.; Yao, K.; Wang, S.; Yin, J.; Ma, X.; Yin, X.; Wang, X.; Sun, Y. Understanding the research advances on lumpy skin disease: A comprehensive literature review of experimental evidence. Front. Microbiol. 2022, 13, 1065894. [Google Scholar] [CrossRef] [PubMed]
- Gari, G.; Abie, G.; Gizaw, D.; Wubete, A.; Kidane, M.; Asgedom, H.; Bayissa, B.; Ayelet, G.; Oura, C.A.; Roger, F. Evaluation of the safety, immunogenicity and efficacy of three capripoxvirus vaccine strains against lumpy skin disease virus. Vaccine 2015, 33, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Haegeman, A.; De Leeuw, I.; Mostin, L.; Campe, W.V.; Aerts, L.; Venter, E.; Tuppurainen, E.; Saegerman, C.; De Clercq, K. Comparative evaluation of lumpy skin disease virus-based live attenuated vaccines. Vaccines 2021, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Klement, E.; Broglia, A.; Antoniou, S.-E.; Tsiamadis, V.; Plevraki, E.; Petrović, T.; Polaček, V.; Debeljak, Z.; Miteva, A.; Alexandrov, T. Neethling vaccine proved highly effective in controlling lumpy skin disease epidemics in the Balkans. Prev. Vet. Med. 2020, 181, 104595. [Google Scholar] [CrossRef] [PubMed]
- HHamdi, J.; Boumart, Z.; Daouam, S.; El Arkam, A.; Bamouh, Z.; Jazouli, M.; Tadlaoui, K.O.; Fihri, O.F.; Gavrilov, B.; El Harrak, M. Development and evaluation of an inactivated lumpy skin disease vaccine for cattle. Vet. Microbiol. 2020, 245, 108689. [Google Scholar]
- Es-Sadeqy, Y.; Bamouh, Z.; Ennahli, A.; Safini, N.; El Mejdoub, S.; Tadlaoui, K.O.; Gavrilov, B.; El Harrak, M. Development of an inactivated combined vaccine for protection of cattle against lumpy skin disease and bluetongue viruses. Vet. Microbiol. 2021, 256, 109046. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Moritz, T.; Schlottau, K.; Hoffmann, D.; Beer, M.; Hoffmann, B. Development of a safe and highly efficient inactivated vaccine candidate against lumpy skin disease virus. Vaccines 2020, 9, 4. [Google Scholar] [CrossRef]
- Mazloum, A.; Van Schalkwyk, A.; Babiuk, S.; Venter, E.; Wallace, D.B.; Sprygin, A. Lumpy skin disease: History, current understanding and research gaps in the context of recent geographic expansion. Front. Microbiol. 2023, 14, 1266759. [Google Scholar] [CrossRef]
- Tuppurainen, E.; Dietze, K.; Wolff, J.; Bergmann, H.; Beltran-Alcrudo, D.; Fahrion, A.; Lamien, C.E.; Busch, F.; Sauter-Louis, C.; Conraths, F.J.; et al. Vaccines and vaccination against lumpy skin disease. Vaccines 2021, 9, 1136. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Pearson, C.R.; Bachanek-Bankowska, K.; Knowles, N.J.; Amareen, S.; Frost, L.; Henstock, M.R.; Lamien, C.E.; Diallo, A.; Mertens, P.P. Characterization of sheep pox virus vaccine for cattle against lumpy skin disease virus. Antivir. Res. 2014, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bazid, A.-H.; Wasfy, M.; Fawzy, M.; Nayel, M.; Abdelmegeid, M.; Thabet, R.Y.; Yong, H.S.; El-Sayed, M.M.; Magouz, A.; Badr, Y. Emergency vaccination of cattle against lumpy skin disease: Evaluation of safety, efficacy, and potency of MEVAC® LSD vaccine containing Neethling strain. Vet. Res. Commun. 2023, 47, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Barua, S.; Kumar, R.; Khandelwal, N.; Kumar, A.; Verma, A.; Singh, L.; Godara, B.; Chander, Y.; Kumar, G.; et al. Evaluation of the safety, immunogenicity and efficacy of a new live-attenuated lumpy skin disease vaccine in India. Virulence 2023, 14, 2190647. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.; Arif, B.; Black, D.; Dumbell, K.; Esposito, J.; Lefkowitz, E.; McFadden, G.; Moss, B.; Mercer, A.; Moyer, R. Family poxviridae. In Virus Taxonomy: VIIIth Report of the International Committee on Taxonomy of Viruses; Academic Press: Cambridge, MA, USA, 2005; pp. 117–133. [Google Scholar]
- Tulman, E.; Afonso, C.; Lu, Z.; Zsak, L.; Kutish, G.; Rock, D. Genome of lumpy skin disease virus. J. Virol. 2001, 75, 7122–7130. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, G.; Mahrosh, H.S.; Salman, M.; Ali, M.; Arif, R.; Ahmed, S.; Ebaid, H. In Silico Analysis of Honey Bee Peptides as Potential Inhibitors of Capripoxvirus DNA-Directed RNA Polymerase. Animals 2023, 13, 2281. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wang, H.; Wang, Z.; Wang, Q. Progression of Antiviral Agents Targeting Viral Polymerases. Molecules 2022, 27, 7370. [Google Scholar] [CrossRef]
- Akther, M.; Akter, S.H.; Sarker, S.; Aleri, J.W.; Annandale, H.; Abraham, S.; Uddin, J.M. Global Burden of Lumpy Skin Disease, Outbreaks, and Future Challenges. Viruses 2023, 15, 1861. [Google Scholar] [CrossRef]
- Yadav, D.; Rao, G.S.; Paliwal, D.; Singh, A.; Alam, A.; Kumar Sharma, P.; Surendra, A.V.; Varshney, P.; Kumar, Y. Cracking the Code of Lumpy Skin Disease: Identifying Causes, Symptoms and Treatment Options for Livestock Farmers. Infect. Disord.-Drug Targets (Former. Curr. Drug Targets-Infect. Disord.) 2024, 24, 57–71. [Google Scholar] [CrossRef]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef]
- Magden, J.; Kääriäinen, L.; Ahola, T. Inhibitors of virus replication: Recent developments and prospects. Appl. Microbiol. Biotechnol. 2005, 66, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Vardhan, S.; Sahoo, S.K. Computational studies on searching potential phytochemicals against DNA polymerase activity of the monkeypox virus. J. Tradit. Complement. Med. 2023, 13, 465–478. [Google Scholar] [CrossRef]
- Sharma, R.; Bhattu, M.; Tripathi, A.; Verma, M.; Acevedo, R.; Kumar, P.; Rajput, V.D.; Singh, J. Potential medicinal plants to combat viral infections: A way forward to environmental biotechnology. Environ. Res. 2023, 227, 115725. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, J.; Mohan, M.; Byrareddy, S.N. Drug repurposing approaches to combating viral infections. J. Clin. Med. 2020, 9, 3777. [Google Scholar] [CrossRef]
- Mercorelli, B.; Palù, G.; Loregian, A. Drug repurposing for viral infectious diseases: How far are we? Trends Microbiol. 2018, 26, 865–876. [Google Scholar] [CrossRef]
- Punekar, M.; Kshirsagar, M.; Tellapragada, C.; Patil, K. Repurposing of antiviral drugs for COVID-19 and impact of repurposed drugs on the nervous system. Microb. Pathog. 2022, 168, 105608. [Google Scholar] [CrossRef]
- Toker, E.B.; Ates, O.; Yeşilbağ, K. Inhibition of bovine and ovine capripoxviruses (Lumpy skin disease virus and Sheeppox virus) by ivermectin occurs at different stages of propagation in vitro. Virus Res. 2022, 310, 198671. [Google Scholar] [CrossRef]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Sciortino, M.T. Antiviral activity exerted by natural products against human viruses. Viruses 2021, 13, 828. [Google Scholar] [CrossRef] [PubMed]
- Badhy, S.C.; Chowdhury, M.G.A.; Settypalli, T.B.K.; Cattoli, G.; Lamien, C.E.; Fakir, M.A.U.; Akter, S.; Osmani, M.G.; Talukdar, F.; Begum, N. Molecular characterization of lumpy skin disease virus (LSDV) emerged in Bangladesh reveals unique genetic features compared to contemporary field strains. BMC Vet. Res. 2021, 17, 1–11. [Google Scholar] [CrossRef]
- Cramer, P. AlphaFold2 and the future of structural biology. Nat. Struct. Mol. Biol. 2021, 28, 704–705. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Agnihotry, S.; Pathak, R.K.; Singh, D.B.; Tiwari, A.; Hussain, I. Protein structure prediction. In Bioinformatics; Academic Press: Cambridge, MA, USA, 2022; pp. 177–188. [Google Scholar]
- Sharma, C.; Nigam, A.; Singh, R. Computational-approach understanding the structure-function prophecy of Fibrinolytic Protease RFEA1 from Bacillus cereus RSA1. PeerJ 2021, 9, e11570. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37 (Suppl. S1), D211–D215. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2021-2: SiteMap; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34 (Suppl. 1), D668–D672. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2023-2: Maestro; Schrödinger, LLC: New York, NY, USA, 2023. [Google Scholar]
- Schrödinger. Schrödinger Release 2023-2: Protein Preparation Wizard Epik; Schrödinger, LLC: New York, NY, USA, 2023. [Google Scholar]
- Schrödinger. Schrödinger Release 2023-3: SiteMap; Schrödinger, LLC: New York, NY, USA, 2023. [Google Scholar]
- Tahlan, S.; Kumar, S.; Ramasamy, K.; Lim, S.M.; Shah, S.A.A.; Mani, V.; Narasimhan, B. In-silico molecular design of heterocyclic benzimidazole scaffolds as prospective anticancer agents. BMC Chem. 2019, 13, 1–22. [Google Scholar] [CrossRef]
- David, T.I.; Adelakun, N.S.; Omotuyi, O.I.; Metibemu, D.S.; Ekun, O.E.; Inyang, O.K.; Adewumi, B.; Enejoh, O.A.; Owolabi, R.T.; Oribamise, E.I. Molecular docking analysis of phyto-constituents from Cannabis sativa with pfDHFR. Bioinformation 2018, 14, 574. [Google Scholar] [CrossRef]
- Singh, N.; Chaput, L.; Villoutreix, B.O. Virtual screening web servers: Designing chemical probes and drug candidates in the cyberspace. Brief. Bioinform. 2021, 22, 1790–1818. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. Schrödinger Release 2019-3: Glide; Schrödinger LLC: New York, NY, USA, 2019. [Google Scholar]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins: Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Kadioglu, O.; Saeed, M.; Greten, H.J.; Efferth, T. Identification of novel compounds against three targets of SARS CoV-2 coronavirus by combined virtual screening and supervised machine learning. Comput. Biol. Med. 2021, 133, 104359. [Google Scholar] [CrossRef]
- Ajjarapu, S.M.; Tiwari, A.; Taj, G.; Singh, D.B.; Singh, S.; Kumar, S. Simulation studies, 3D QSAR and molecular docking on a point mutation of protein kinase B with flavonoids targeting ovarian Cancer. BMC Pharmacol. Toxicol. 2021, 22, 1–23. [Google Scholar] [CrossRef]
- Ghosh, S.; Nie, A.; An, J.; Huang, Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr. Opin. Chem. Biol. 2006, 10, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Pattar, S.V.; Adhoni, S.A.; Kamanavalli, C.M.; Kumbar, S.S. In silico molecular docking studies and MM/GBSA analysis of coumarin-carbonodithioate hybrid derivatives divulge the anticancer potential against breast cancer. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 1–10. [Google Scholar] [CrossRef]
- Sankar, M.; Ramachandran, B.; Pandi, B.; Mutharasappan, N.; Ramasamy, V.; Prabu, P.G.; Shanmugaraj, G.; Wang, Y.; Muniyandai, B.; Rathinasamy, S. In silico screening of natural phytocompounds towards identification of potential lead compounds to treat COVID-19. Front. Mol. Biosci. 2021, 8, 637122. [Google Scholar] [CrossRef]
- Sun, T.-Y.; Wang, Q.; Zhang, J.; Wu, T.; Zhang, F. Trastuzumab-Peptide interactions: Mechanism and application in structure-based ligand design. Int. J. Mol. Sci. 2013, 14, 16836–16850. [Google Scholar] [CrossRef]
- Schrödinger. QikProp. In Schrödinger Release 2021-2; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K. Bringing molecular dynamics simulation data into view. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of lead compounds against Scm (fms10) in Enterococcus faecium using computer aided drug designing. Life 2021, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Chávez, R.M.; Salerno, A.; Liuzzi, A.; Ilari, A.; Milelli, A.; Uliassi, E.; Bolognesi, M.L. Targeted Protein Degradation for Infectious Diseases: From Basic Biology to Drug Discovery. ACS Bio Med Chem Au 2022, 3, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Kara, P.; Afonso, C.; Wallace, D.; Kutish, G.; Abolnik, C.; Lu, Z.; Vreede, F.; Taljaard, L.; Zsak, A.; Viljoen, G.J. Comparative sequence analysis of the South African vaccine strain and two virulent field isolates of lumpy skin disease virus. Arch. Virol. 2003, 148, 1335–1356. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, M.; Bebenek, K. Multiple functions of DNA polymerases. Crit. Rev. Plant Sci. 2007, 26, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Alzyoud, L.; Bryce, R.A.; Al Sorkhy, M.; Atatreh, N.; Ghattas, M.A. Structure-based assessment and druggability classification of protein–protein interaction sites. Sci. Rep. 2022, 12, 7975. [Google Scholar] [CrossRef]
- Ali, M.C.; Nur, A.J.; Khatun, M.S.; Dash, R.; Rahman, M.M.; Karim, M.M. Identification of potential SARS-CoV-2 main protease inhibitors from Ficus Carica Latex: An in-silico approach. J. Adv. Biotechnol. Exp. Ther. 2020, 3, 57–67. [Google Scholar] [CrossRef]
- Sezer, A.; Halilović-Alihodžić, M.; Vanwieren, A.R.; Smajkan, A.; Karić, A.; Djedović, H.; Šutković, J. A review on drug repurposing in COVID-19: From antiviral drugs to herbal alternatives. J. Genet. Eng. Biotechnol. 2022, 20, 78. [Google Scholar] [CrossRef]
- Mohanty, S.S.; Sahoo, C.R.; Paidesetty, S.K.; Padhy, R.N. Role of phytocompounds as the potential anti-viral agent: An overview. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 2311–2329. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen Bonds in Proteins: Role and Strength. In Encyclopedia of Life Science; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Wade, R.C.; Goodford, P.J. The role of hydrogen-bonds in drug binding. Prog. Clin. Biol. Res. 1989, 289, 433–444. [Google Scholar] [PubMed]
- Boehr, D.D.; Farley, A.R.; Wright, G.D.; Cox, J.R. Analysis of the π-π stacking interactions between the aminoglycoside antibiotic kinase APH (3′)-IIIa and its nucleotide ligands. Chem. Biol. 2002, 9, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Spassov, D.S.; Atanasova, M.; Doytchinova, I. A role of salt bridges in mediating drug potency: A lesson from the N-myristoyltransferase inhibitors. Front. Mol. Biosci. 2023, 9, 1066029. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. Cation-π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Challapa-Mamani, M.R.; Tomás-Alvarado, E.; Espinoza-Baigorria, A.; León-Figueroa, D.A.; Sah, R.; Rodriguez-Morales, A.J.; Barboza, J.J. Molecular Docking and Molecular Dynamics Simulations in Related to Leishmania donovani: An Update and Literature Review. Trop. Med. Infect. Dis. 2023, 8, 457. [Google Scholar] [CrossRef] [PubMed]
- Munni, Y.A.; Ali, M.C.; Selsi, N.J.; Sultana, M.; Hossen, M.; Bipasha, T.H.; Rahman, M.; Uddin, M.N.; Hosen, S.Z.; Dash, R. Molecular simulation studies to reveal the binding mechanisms of shikonin derivatives inhibiting VEGFR-2 kinase. Comput. Biol. Chem. 2021, 90, 107414. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, M.A.; Basheera, S.; Sivanandan, S. Inhibition of Monkeypox Virus DNA Polymerase Using Moringa oleifera Phytochemicals: Computational Studies of Drug-Likeness, Molecular Docking, Molecular Dynamics Simulation and Density Functional Theory. Indian J. Microbiol. 2024, 1–18. [Google Scholar] [CrossRef]
- Liang, J.J.; Pitsillou, E.; Hung, A.; Karagiannis, T.C. A repository of COVID-19 related molecular dynamics simulations and utilisation in the context of nsp10-nsp16 antivirals. J. Mol. Graph. Model. 2024, 126, 108666. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ji, J.; Zhong, Y.; Meng, W.; Wan, S.; Ding, X.; Chen, Z.; Wu, W.; Jia, K.; Li, S. Construction of recombinant fluorescent LSDV for high-throughput screening of antiviral drugs. Vet. Res. 2024, 55, 1–14. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, X.; Guo, A.; Zhang, S.; Zou, Y.; Wang, Y.; Li, X.; He, W.; Pu, L.; Zhang, S.; et al. HIV protease inhibitor nelfinavir is a potent drug candidate against echinococcosis by targeting Ddi1-like protein. EBioMedicine 2022, 82, 104177. [Google Scholar] [CrossRef]
- Mabrouk Gabr, N.; Hassan Mohammed, I. A comparative study of canagliflozin (INVOKANA) on type-I and type-II diabetes mellitus on adult male albino rat. Al-Azhar Med. J. 2020, 49, 15–32. [Google Scholar] [CrossRef]
- Deeks, E.D.; Scheen, A.J. Canagliflozin: A review in type 2 diabetes. Drugs 2017, 77, 1577–1592. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.E.; Cersosimo, E. Use of canagliflozin in combination with and compared to incretin-based therapies in type 2 diabetes. Clin. Diabetes 2017, 35, 141–153. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Roskoski Jr, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Javed, S. Elucidation of binding dynamics of tyrosine kinase inhibitor tepotinib, to human serum albumin, using spectroscopic and computational approach. Int. J. Biol. Macromol. 2023, 241, 124656. [Google Scholar] [CrossRef] [PubMed]
- Abduljalil, J.M.; Elfiky, A.A.; AlKhazindar, M.M. Tepotinib and tivantinib as potential inhibitors for the serine/threonine kinase of the mpox virus: Insights from structural bioinformatics analysis. J. Biomol. Struct. Dyn. 2024, 1–11. [Google Scholar] [CrossRef]
- Vijayasri, S.; Hopper, W. Towards the identification of novel phytochemical leads as macrodomain inhibitors of chikungunya virus using molecular docking approach. J. Appl. Pharm. Sci. 2017, 7, 074–082. [Google Scholar]
- Siddiquee, N.H.; Malek, S.; Tanni, A.A.; Mitu, I.J.; Arpa, S.H.; Hasan, M.R.; Shammi, S.E.; Chakma, C.; Mahinur, M.; Wajed, S.; et al. Unveiling the antiviral activity of 2′, 3, 5, 7-Tetrahydroxyflavanone as potential inhibitor of chikungunya virus envelope glycoprotein. Inform. Med. Unlocked 2024, 47, 101486. [Google Scholar] [CrossRef]
- Luczkowiak, J.; Álvarez, M.; Sebastián-Martín, A.; Menéndez-Arias, L. DNA-dependent DNA polymerases as drug targets in herpesviruses and poxviruses. Viral Polym. 2019, 95–134. [Google Scholar] [CrossRef]
- Kumari, K.; Kumar, A.; Singh, P.; Kaushik, N.K. In silico study of remdesivir with and without ionic liquids having different cations using DFT calculations and molecular docking. J. Indian Chem. Soc. 2022, 99, 100328. [Google Scholar] [CrossRef]
- Murgueitio, M.S.; Bermudez, M.; Mortier, J.; Wolber, G. In silico virtual screening approaches for anti-viral drug discovery. Drug Discov. Today Technol. 2012, 9, e219–e225. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Shahin, F.; Zhai, W.; Li, H.; Alvisi, G.; Yang, K.; Chen, X.; Chen, Y.; Chen, J.; Hu, C.; et al. Ivermectin inhibits bovine herpesvirus 1 DNA polymerase nuclear import and interferes with viral replication. Microorganisms 2020, 8, 409. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Chakraborty, S.; Ahmad, M.; Kumar, V.; Tailor, P.B.; Biswal, B.K. Identification of probable inhibitors for the DNA polymerase of the Monkeypox virus through the virtual screening approach. Int. J. Biol. Macromol. 2023, 229, 515–528. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Binding Site Residues |
|---|---|
| Lumpy skin disease virus-encoded DNA polymerase (LSDV 039) | Lys 4, Glu 122, Gly 123, Cys 124, Arg 155, Phe156, Asn 157, Ile 158, Asn 159, Arg 160, Tyr 162, Phe 164, Ile 191, Asn 195, Leu 304, Phe 329, Thr 333, Tyr 334, Lys 337, Ser 338, Glu 339, Lys 340, Asn 352, Ala 353, Phe 354, Ser 355, Cys 356, Asn 374, Ile 379, Gly 380, Lys 381, Ile 382, Ser 383, Ser 384, Phe 385, Glu 387, Val 388, Asp 412, Tyr 473, Trp 475, Asn 476, Tyr 477, Tyr 478, Gly 479, Ile 480, Glu 481, Thr 482, Lys 483, Asp 485, Ala 486, Gly 487, Phe 489, Tyr 491, Val 498, Phe 499, Glu 500, Tyr 501, Arg 502, Ala 503, Leu 506, Tyr642, Tyr646, Leu 651, Ser 652, Thr 653, Lys 655, Ser 656, Ile 657, Tyr 658, Asn 659, Ser 660, Met 661, Glu 662, Tyr 663, Thr 664, Tyr 665, Ile 667, Ile 668, Ser 671 |
| Gene | Uniprot ID | Site Map Analysis | Errat Value | Ramachandran Plot | |||
|---|---|---|---|---|---|---|---|
| Most Favored Regions | Additional Allowed Regions | Generously Allowed Regions | Disallowed Region | ||||
| LSDV039 | Q91MW8 | 1.048 | 88.8 | 91% | 8.0% | 0.6% | 0.3% |
| Compound Type | Compound ID | Name | Glide Gscore (kcal/mol) | Glide Emodel (kcal/mol) | MMGBSA dGbind (kcal/mol) |
|---|---|---|---|---|---|
| Positive control Antiviral drugs | CID6321424 | Ivermectin B1a | −5.63 | −67.89 | −26.28 |
| DB08907 | Canagliflozin | −9.86 | −69.37 | −45.68 | |
| DB15133 | Tepotinib | −8.86 | −88.69 | −47.99 | |
| Phytocompounds | CID 10168 | Rhein | −8.97 | −43.08 | −44.72 |
| CID 439533 | Taxifolin | −7.20 | −51.87 | −44.48 |
| Compound Type | Compound ID | Name | Residues in Interaction | Bond Distance (Å) | Bond Type |
|---|---|---|---|---|---|
| Positive control | CID6321424 | Ivermectin B1a | Asp 337 Ser 338 Glu 339 | 1.83 2.16 1.55 | H-bond H-bond H-bond |
| Antiviral drugs | DB08907 | Canagliflozin | Lys 483 Asn 659 Asn 659 | 2.61 1.80 1.73 | Salt bridge H-bond H-bond |
| DB15133 | Tepotinib | Glu 339 Glu 339 Tyr 477 Lys 483 Phe 499 Tyr 663 | 1.93 2.94 5.49 4.01 4.89 5.38 | H-bond Salt bridge Pi–pi stacking Pi–cation Pi–pi stacking Pi–pi stacking | |
| Phytocompounds | CID 10168 | Rhein | Lys 337 Val 498 Thr 664 | 2.71 2.12 2.09 | Salt bridge H-bond H-bond |
| CID 439533 | Taxifolin | Ser 338 Glu 339 Lys 483 Lys 483 Ser 656 Thr 664 | 2.10 1.78 2.00 3.90 2.02 2.05 | H-bond H-bond H-bond Pi–cation H-bond H-bond |
| Compound Type | Compound ID | Name | ∆G Bind (NS) | ∆G Coulomb (NS) | ∆G Lipo (NS) | ∆G vdW (NS) |
|---|---|---|---|---|---|---|
| Positive control Antiviral drugs | CID6321424 | Ivermectin B1a | −39.70 | −15.52 | −17.01 | −63.90 |
| DB08907 | Canagliflozin | −51.44 | −46.81 | −25.21 | −42.95 | |
| DB15133 | Tepotinib | −54.06 | 50.09 | −23.28 | −58.20 | |
| Phytocompounds | CID 10168 | Rhein | −38.20 | −77.35 | −13.22 | −34.55 |
| CID 439533 | Taxifolin | −47.80 | −29.02 | −13.33 | −24.43 |
| Compound Type | Name | #Star 1 | Molecular Weight 2 | SASA 3 | FISA 4 | QPlogPo/w 5 | QPlogS 6 | QPlogBB 7 | QPlog HERG 8 | QPlogKp 9 | Percent Human Oral Absorption 10 | Rule of Five 11 | Rule of Tree 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive control | Ivermectin B1a | 11 | 875.10 | 1232.66 | 137.09 | 6.398 | −8.09 | −2.28 | −5.98 | −2.11 | 73.19 | 3 | 2 |
| Antiviral drugs | Canagliflozin | 4 | 438.47 | 693.64 | 174.33 | 4.01 | −6.14 | −1.35 | −6.41 | −2.97 | 92.33 | 0 | 2 |
| Tepotinib | 1 | 494.60 | 881.97 | 94.45 | 4.59 | −6.75 | −0.55 | −8.58 | −3.17 | 100.00 | 0 | 2 | |
| Phytocompounds | Rhein | 0 | 284.23 | 478.12 | 271.67 | 0.98 | −2.66 | −1.97 | −2.69 | −5.51 | 47.41 | 0 | 1 |
| Taxifolin | 0 | 304.26 | 518.52 | 276.85 | 0.11 | −2.73 | −2.27 | −4.928 | −5.38 | 52.10 | 0 | 0 |
| System | RMSD (Å) | RMSF (Å) | Rg (Å) | SASA (Å) |
|---|---|---|---|---|
| Ivermectin B1a | 0.88 | 1.58 | 32.72 | 47,186.72 |
| Canagliflozin | 0.88 | 1.69 | 32.97 | 47,677.70 |
| Tepotinib | 1.44 | 1.26 | 32.99 | 47,724.08 |
| Rhein | 0.33 | 1.62 | 33.18 | 48,127.33 |
| Taxifolin | 0.51 | 1.63 | 32.89 | 47,439.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zia, S.; Sumon, M.-M.; Ashik, M.-A.; Basar, A.; Lim, S.; Oh, Y.; Park, Y.; Rahman, M.-M. Potential Inhibitors of Lumpy Skin Disease’s Viral Protein (DNA Polymerase): A Combination of Bioinformatics Approaches. Animals 2024, 14, 1283. https://doi.org/10.3390/ani14091283
Zia S, Sumon M-M, Ashik M-A, Basar A, Lim S, Oh Y, Park Y, Rahman M-M. Potential Inhibitors of Lumpy Skin Disease’s Viral Protein (DNA Polymerase): A Combination of Bioinformatics Approaches. Animals. 2024; 14(9):1283. https://doi.org/10.3390/ani14091283
Chicago/Turabian StyleZia, Sabbir, Md-Mehedi Sumon, Md-Ashiqur Ashik, Abul Basar, Sangjin Lim, Yeonsu Oh, Yungchul Park, and Md-Mafizur Rahman. 2024. "Potential Inhibitors of Lumpy Skin Disease’s Viral Protein (DNA Polymerase): A Combination of Bioinformatics Approaches" Animals 14, no. 9: 1283. https://doi.org/10.3390/ani14091283