Genome-Wide Scan Identifies Selection Signatures in Chinese Wagyu Cattle Using a High-Density SNP Array

Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Ethics Statement
2.2. Genotypes
2.3. Detection of Selection Signatures Using iHS
2.4. Bioinformatics Analyses of Candidate Genes Under Selection
3. Results
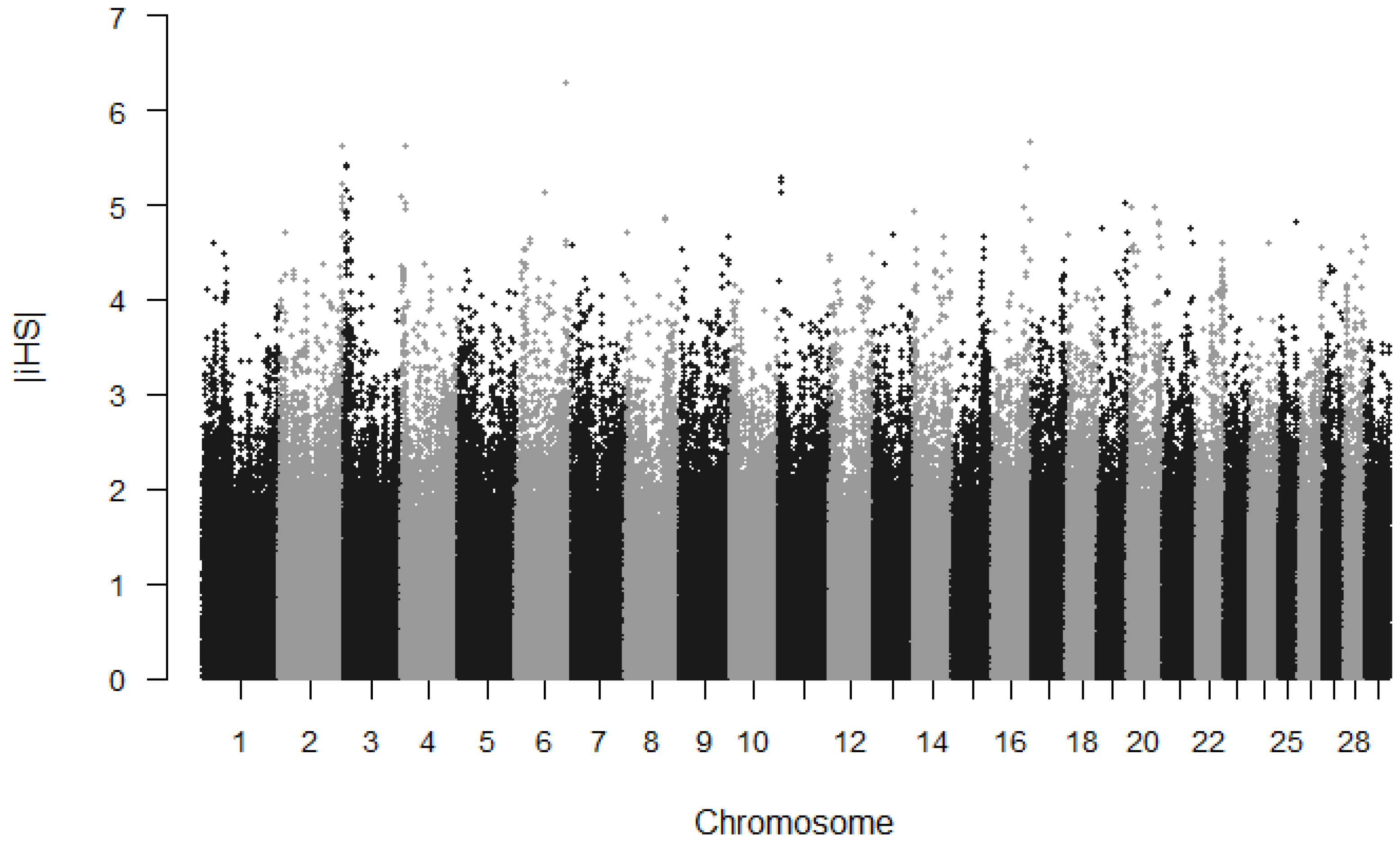
3.1. Detection of Selection Signature Using an iHS Approach
3.2. Genes and QTLs Based on Identified Regions
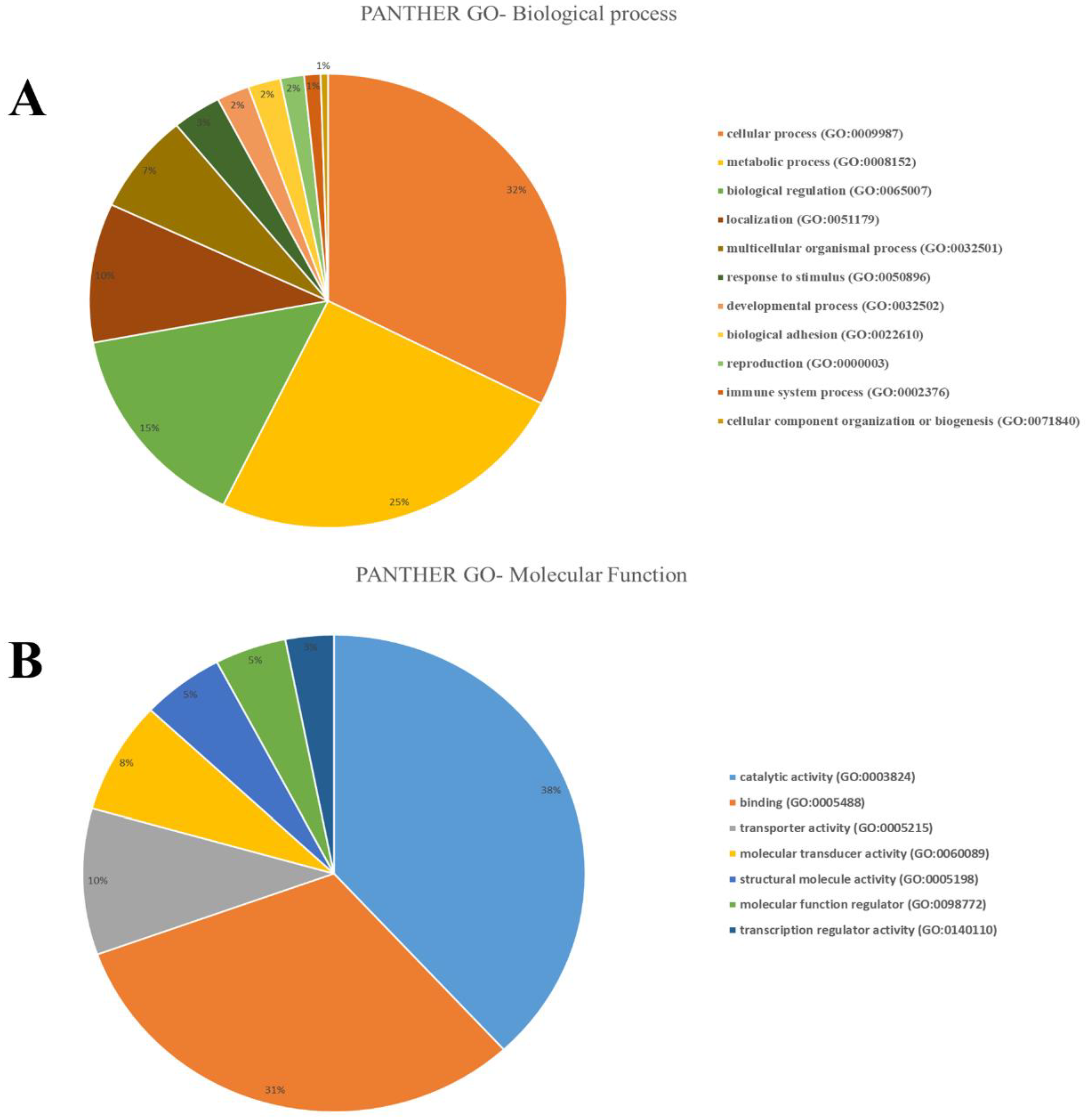
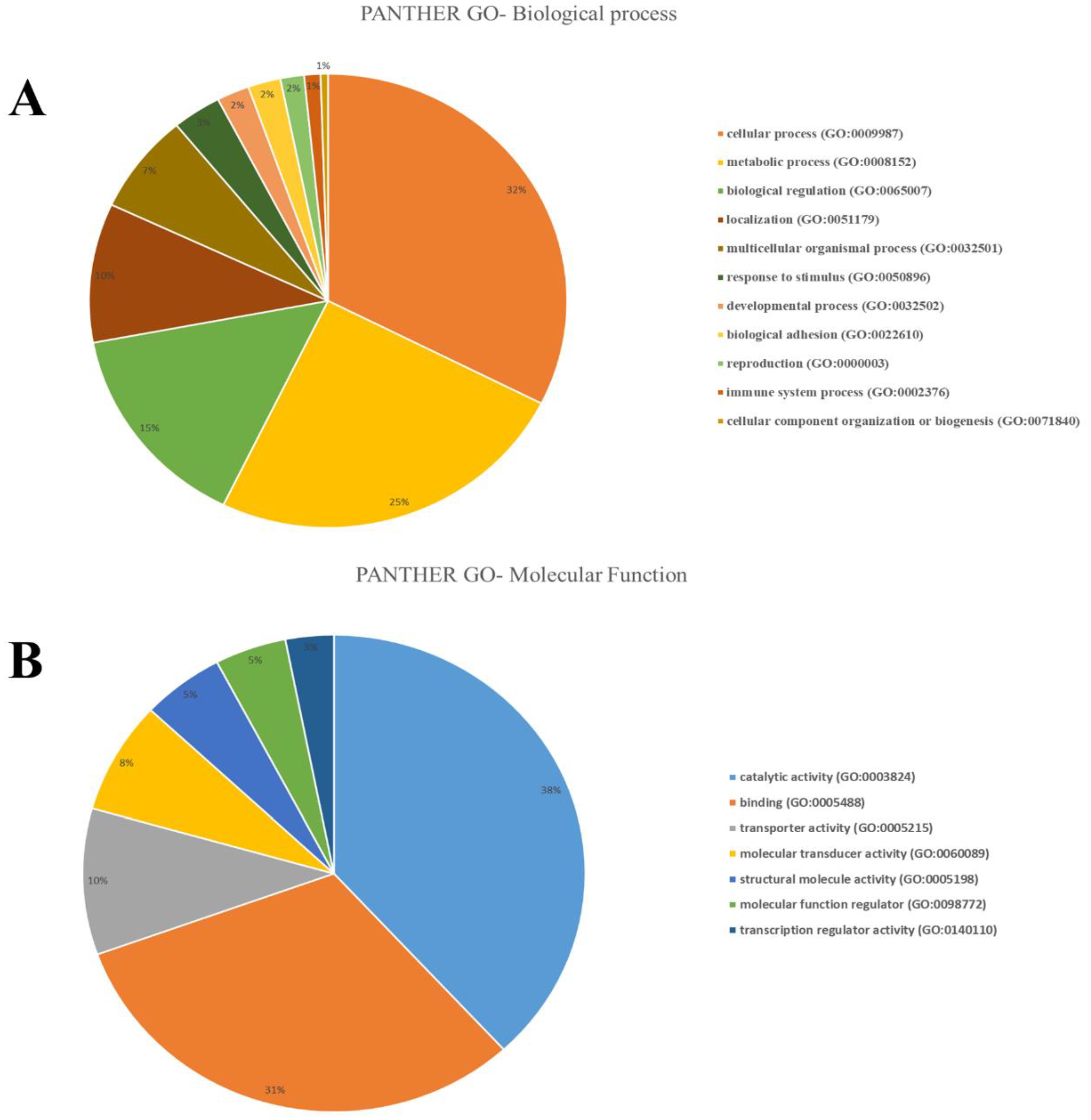
3.3. Functional Classification based on Selection Genes
3.4. Candidate Genes under Selection
4. Discussion
4.1. Candidate Genes Related to Meat Quality
4.2. Genes Related to Fatty Acids
4.3. Genes Related to Growth and Developmental Performance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akey, J.M.; Zhang, G.; Zhang, K.; Jin, L.; Shriver, M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002, 419, 832–837. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Admetlla, A.; Liang, M.; Korneliussen, T.; Nielsen, R. On Detecting Incomplete Soft or Hard Selective Sweeps Using Haplotype Structure. Mol. Biol. Evol. 2014, 31, 1275–1291. [Google Scholar] [CrossRef] [Green Version]
- Qanbari, S.; Gianola, D.; Hayes, B.; Schenkel, F.; Miller, S.; Moore, S.; Thaller, G.; Simianer, H. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genom. 2011, 12, 318. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.C.; Sabeti, P.; Hutcheson, H.B.; Fry, B.; Schaffner, S.F.; de Bakker, P.I.W.; Varilly, P.; Palma, A.A.; Roy, J.; Cooper, R.; et al. Searching for signals of evolutionary selection in 168 genes related to immune function. Hum. Genet. 2006, 119, 92–102. [Google Scholar] [CrossRef]
- Pybus, M.; Dall Olio, G.M.; Luisi, P.; Uzkudun, M.; Carreño-Torres, A.; Pavlidis, P.; Laayouni, H.; Bertranpetit, J.; Engelken, J. 1000 Genomes Selection Browser 1.0: A genome browser dedicated to signatures of natural selection in modern humans. Nucleic Acids Res. 2013, 42, D903–D909. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Akey, J.M. Genomic insights into positive selection. Trends Genet. 2006, 22, 437–446. [Google Scholar] [CrossRef]
- Grossman, S.R.; Shylakhter, I.; Karlsson, E.K.; Byrne, E.H.; Morales, S.; Frieden, G.; Hostetter, E.; Angelino, E.; Garber, M.; Zuk, O.; et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 2010, 327, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, H.; Zhang, Q.; Ding, X. Identification of selection footprints on the X chromosome in pig. PLoS ONE 2014, 9, e94911. [Google Scholar] [CrossRef]
- Stella, A.; Ajmone-Marsan, P.; Lazzari, B.; Boettcher, P. Identification of Selection Signatures in Cattle Breeds Selected for Dairy Production. Genetics 2010, 185, 1451–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qanbari, S.; Pimentel, E.C.G.; Tetens, J.; Thaller, G.; Lichtner, P.; Sharifi, A.R.; Simianer, H. A genome-wide scan for signatures of recent selection in Holstein cattle. Anim. Genet. 2010, 41, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Chamberlain, A.J.; Maceachern, S.; Savin, K.; McPartlan, H.; MacLeod, I.; Sethuraman, L.; Goddard, M.E. A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim. Genet. 2009, 40, 176–184. [Google Scholar] [CrossRef]
- Gautier, M.; Naves, M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 2011, 20, 3128–3143. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, X.; Li, K.; Fan, B.; Tang, Z. A genome-wide scan for signatures of selection in Chinese indigenous and commercial pig breeds. BMC Genet. 2014, 15, 7. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Sonstegard, T.S.; Liu, G.E.; Bickhart, D.M.; Da Silva, M.V.B.; Machado, M.A.; Utsunomiya, Y.T.; Garcia, J.F.; Gondro, C.; Van Tassell, C.P. Genomic divergence of zebu and taurine cattle identified through high-density SNP genotyping. BMC Genom. 2013, 14, 876. [Google Scholar] [CrossRef] [PubMed]
- Rothammer, S.; Seichter, D.; Förster, M.; Medugorac, I. A genome-wide scan for signatures of differential artificial selection in ten cattle breeds. BMC Genom. 2013, 14, 908. [Google Scholar] [CrossRef]
- Utsunomiya, Y.T.; Pérez O’Brien, A.M.; Sonstegard, T.S.; Van Tassell, C.P.; Do Carmo, A.S.; Mészáros, G.; Sölkner, J.; Garcia, J.F. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS ONE 2013, 8, e64280. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Yamaji, K.; Uemoto, Y.; Sasago, N.; Kobayashi, E.; Kobayashi, N.; Matsuhashi, T.; Maruyama, S.; Matsumoto, H.; Sasazaki, S.; et al. Genome-wide association study for fatty acid composition in Japanese Black cattle. Anim. Sci. J. 2013, 84, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Kobayashi, M.; Shoji, N.; Kato, K. Genetic parameters for fatty acid composition and feed efficiency traits in Japanese Black cattle. Animal 2011, 5, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Nogi, T.; Honda, T.; Mukai, F.; Okagaki, T.; Oyama, K. Heritabilities and genetic correlations of fatty acid compositions in longissimus muscle lipid with carcass traits in Japanese Black cattle. J. Anim. Sci. 2011, 89, 615–621. [Google Scholar] [CrossRef]
- Uemoto, Y.; Abe, T.; Tameoka, N.; Hasebe, H.; Inoue, K.; Nakajima, H.; Shoji, N.; Kobayashi, M.; Kobayashi, E. Whole-genome association study for fatty acid composition of oleic acid in Japanese Black cattle. Anim. Genet. 2011, 42, 141–148. [Google Scholar] [CrossRef]
- Matsuhashi, T.; Maruyama, S.; Uemoto, Y.; Kobayashi, N.; Mannen, H.; Abe, T.; Sakaguchi, S.; Kobayashi, E. Effects of bovine fatty acid synthase, stearoyl-coenzyme A desaturase, sterol regulatory element-binding protein 1, and growth hormone gene polymorphisms on fatty acid composition and carcass traits in Japanese Black cattle. J. Anim. Sci. 2011, 89, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B.; Lunt, D.K.; Chung, K.Y.; Choi, C.B.; Tume, R.K.; Zembayashi, M. Adiposity, fatty acid composition, and delta-9 desaturase activity during growth in beef cattle. Anim. Sci. J. 2006, 77, 478–486. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, B.; Niu, H.; Zhang, W.; Xu, L.; Xu, L.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; et al. Genome wide association study identifies SNPs associated with fatty acid composition in Chinese Wagyu cattle. J. Anim. Sci. Biotechnol. 2019, 10, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpiech, Z.; Hernandez, R. selscan: An efficient multi-threaded program to calculate EHH-based scans for positive selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef]
- Cardoso, D.F.; de Albuquerque, L.G.; Reimer, C.; Qanbari, S.; Erbe, M.; Do Nascimento, A.V.; Venturini, G.C.; Scalez, D.C.B.; Baldi, F.; de Camargo, G.M.F.; et al. Genome-wide scan reveals population stratification and footprints of recent selection in Nelore cattle. Genet. Sel. Evol. 2018, 50, 22. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Haasl, R.J.; Sun, J.; Zhou, Y.; Bickhart, D.M.; Li, J.; Song, J.; Sonstegard, T.S.; Van Tassell, C.P.; Lewin, H.A.; et al. Systematic profiling of short tandem repeats in the cattle genome. Genome Biol. Evol. 2016, 9, 20–31. [Google Scholar] [CrossRef]
- Karolchik, D. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]
- Wang, K.; Wu, P.; Yang, Q.; Chen, D.; Zhou, J.; Jiang, A.; Ma, J.; Tang, Q.; Xiao, W.; Jiang, Y.; et al. Detection of Selection Signatures in Chinese Landrace and Yorkshire Pigs Based on Genotyping-by-Sequencing Data. Front. Genet. 2018, 9, 119. [Google Scholar] [CrossRef]
- Kahi, A.K.; Hirooka, H. Genetic and economic evaluation of Japanese Black (Wagyu) cattle breeding schemes. J. Anim. Sci. 2005, 83, 2021–2032. [Google Scholar] [CrossRef]
- Lloyd, S.S.; Valenzuela, J.L.; Steele, E.J.; Dawkins, R.L. Genetics of Marbling in Wagyu Revealed by the Melting Temperature of Intramuscular and Subcutaneous Lipids. Int. J. Food Sci. 2017, 2017, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagali, N.; Catalá, A. The effect of melatonin and structural analogues on the lipid peroxidation of triglycerides enriched in ω-3 polyunsaturated fatty acids. Life Sci. 2007, 81, 299–305. [Google Scholar] [CrossRef]
- De Oliveira, P.S.N.; Cesar, A.S.M.; Do Nascimento, M.L.; Chaves, A.S.; Tizioto, P.C.; Tullio, R.R.; Lanna, D.P.D.; Rosa, A.N.; Sonstegard, T.S.; Mourao, G.B.; et al. Identification of genomic regions associated with feed efficiency in Nelore cattle. BMC Genet. 2014, 15, 100. [Google Scholar] [CrossRef]
- Gurgul, A.; Szmatoła, T.; Ropka-Molik, K.; Jasielczuk, I.; Pawlina, K.; Semik, E.; Bugno-Poniewierska, M. Identification of genome-wide selection signatures in the Limousin beef cattle breed. J. Anim. Breed. Genet. 2016, 133, 264–276. [Google Scholar] [CrossRef]
- Sevane, N.; Armstrong, E.; Wiener, P.; Pong Wong, R.; Dunner, S. Polymorphisms in twelve candidate genes are associated with growth, muscle lipid profile and meat quality traits in eleven European cattle breeds. Mol. Biol. Rep. 2014, 41, 4721–4731. [Google Scholar] [CrossRef]
- Cheong, H.S.; Yoon, D.; Park, B.L.; Kim, L.H.; Bae, J.S.; Namgoong, S.; Lee, H.W.; Han, C.S.; Kim, J.O.; Cheong, I.; et al. A single nucleotide polymorphism in CAPN1 associated with marbling score in Korean cattle. BMC Genet. 2008, 9, 33. [Google Scholar] [CrossRef]
- Page, B.T.; Casas, E.; Quaas, R.L.; Thallman, R.M.; Wheeler, T.L.; Shackelford, S.D.; Koohmaraie, M.; White, S.N.; Bennett, G.L.; Keele, J.W.; et al. Association of markers in the bovine CAPN1 gene with meat tenderness in large crossbred populations that sample influential industry sires. J. Anim. Sci. 2004, 82, 3474–3481. [Google Scholar] [CrossRef]
- White, S.N.; Casas, E.; Wheeler, T.L.; Shackelford, S.D.; Koohmaraie, M.; Riley, D.G.; Chase, C.C.; Johnson, D.D.; Keele, J.W.; Smith, T.P.L. A new single nucleotide polymorphism in CAPN1 extends the current tenderness marker test to include cattle of Bos indicus, Bos taurus, and crossbred descent1. J. Anim. Sci. 2005, 83, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Casas, E.; White, S.N.; Wheeler, T.L.; Shackelford, S.D.; Koohmaraie, M.; Riley, D.G.; Chase, C.J.; Johnson, D.D.; Smith, T.P. Effects of calpastatin and micro-calpain markers in beef cattle on tenderness traits. J. Anim. Sci. 2006, 84, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A.; Cullen, N.G.; Hickey, S.M.; Dobbie, P.M.; Veenvliet, B.A.; Manley, T.R.; Pitchford, W.S.; Kruk, Z.A.; Bottema, C.D.K.; Wilson, T. Genotypic effects of calpain 1 and calpastatin on the tenderness of cooked M. longissimus dorsi steaks from Jersey × Limousin, Angus and Hereford-cross cattle. Anim. Genet. 2006, 37, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Shi, M. Association analysis of CAPN1 gene variants with carcass and meat quality traits in Chinese native cattle. Afr. J. Biotechnol. 2011, 10, 17367–17371. [Google Scholar]
- Pinto, L.F.B.; Ferraz, J.B.S.; Pedrosa, V.B.; Eler, J.P.; Meirelles, F.V.; Bonin, M.N.; Rezende, F.M.; Carvalho, M.E.; Cucco, D.C.; Silva, R.C.G. Single nucleotide polymorphisms in CAPN and leptin genes associated with meat color and tenderness in Nellore cattle. Genet. Mol. Res. 2011, 10, 2057–2064. [Google Scholar] [CrossRef] [Green Version]
- Xin, J.; Li-chun, Z.; Zhao-zhi, L.; Xiao-hui, L.; Hai-guo, J.; Chang-guo, Y. Association of Polymorphisms in the Calpain I Gene with Meat Quality Traits in Yanbian Yellow Cattle of China. Asian-Australas. J. Anim. Sci. 2011, 24, 9–16. [Google Scholar] [CrossRef]
- Tizioto, P.C.; Decker, J.E.; Taylor, J.F.; Schnabel, R.D.; Mudadu, M.A.; Silva, F.L.; Mourão, G.B.; Coutinho, L.L.; Tholon, P.; Sonstegard, T.S.; et al. Genome scan for meat quality traits in Nelore beef cattle. Physiol. Genom. 2013, 45, 1012–1020. [Google Scholar] [CrossRef] [Green Version]
- Chambaz, A.; Scheeder, M.R.L.; Kreuzer, M.; Dufey, P.A. Meat quality of Angus, Simmental, Charolais and Limousin steers compared at the same intramuscular fat content. Meat Sci. 2003, 63, 491–500. [Google Scholar] [CrossRef]
- Xie, X.; Meng, Q.; Cui, Z.; Ren, L. Effect of Cattle Breed on Meat Quality, Muscle Fiber Characteristics, Lipid Oxidation and Fatty Acids in China. Asian-Australas. J. Anim. Sci. 2012, 25, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Da, C.A.; Bessa, R.J.; Pires, V.M.; Rolo, E.A.; Pinto, R.M.; Andrade, F.C.; Prates, J.A. Is hepatic lipid metabolism of beef cattle influenced by breed and dietary silage level? BMC Vet. Res. 2014, 10, 65. [Google Scholar]
- Lemos, M.V.A.; Chiaia, H.L.J.; Berton, M.P.; Feitosa, F.L.B.; Aboujaoud, C.; Camargo, G.M.F.; Pereira, A.S.C.; Albuquerque, L.G.; Ferrinho, A.M.; Mueller, L.F.; et al. Genome-wide association between single nucleotide polymorphisms with beef fatty acid profile in Nellore cattle using the single step procedure. BMC Genom. 2016, 17, 213. [Google Scholar] [CrossRef]
- Dunner, S.; Sevane, N.; García, D.; Cortés, O.; Valentini, A.; Williams, J.L.; Mangin, B.; Cañón, J.; Levéziel, H. Association of genes involved in carcass and meat quality traits in 15 European bovine breeds. Livest. Sci. 2013, 154, 34–44. [Google Scholar] [CrossRef]
- Perez, R.; Canon, J.; Dunner, S. Genes associated with long-chain omega-3 fatty acids in bovine skeletal muscle. J. Appl. Genet. 2010, 51, 479–487. [Google Scholar] [CrossRef]
- Bernard, C.; Cassar-Malek, I.; Le Cunff, M.; Dubroeucq, H.; Renand, G.; Hocquette, J. New Indicators of Beef Sensory Quality Revealed by Expression of Specific Genes. J. Agric. Food Chem. 2007, 55, 5229–5237. [Google Scholar] [CrossRef] [Green Version]
- Berkowicz, E.W.; Magee, D.A.; Berry, D.P.; Sikora, K.M.; Howard, D.J.; Mullen, M.P.; Evans, R.D.; Spillane, C.; MacHugh, D.E. Single nucleotide polymorphisms in the imprinted bovine insulin-like growth factor 2 receptor gene (IGF2R) are associated with body size traits in Irish Holstein-Friesian cattle. Anim. Genet. 2012, 43, 81–87. [Google Scholar] [CrossRef]
- Petersen, J.L.; Mickelson, J.R.; Rendahl, A.K.; Valberg, S.J.; Andersson, L.S.; Axelsson, J.; Bailey, E.; Bannasch, D.; Binns, M.M.; Borges, A.S.; et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013, 9, e1003211. [Google Scholar] [CrossRef]
- Lettre, G.; Jackson, A.U.; Gieger, C.; Schumacher, F.R.; Berndt, S.I.; Sanna, S.; Eyheramendy, S.; Voight, B.F.; Butler, J.L.; Guiducci, C.; et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008, 40, 584–591. [Google Scholar] [CrossRef]
- Nei, M.; Hughes, A.L. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 1988, 335, 167–170. [Google Scholar]
- Kemper, K.E.; Saxton, S.J.; Bolormaa, S.; Hayes, B.J.; Goddard, M.E. Selection for complex traits leaves little or no classic signatures of selection. BMC Genom. 2014, 15, 246. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| BTA 1 | Position (Mbp) | NO. of SNP 2 (n) | Mean |iHS| Value | Candidate Genes | Gene Name |
|---|---|---|---|---|---|
| 2 | 30.1–30.2 | 22 | 2.198 | SCN9A | Bos taurus sodium voltage-gated channel alpha subunit 9 |
| 2 | 134.4–134.5 | 34 | 2.098 | TAS1R2 | Bos taurus taste 1 receptor member 2 |
| 3 | 14.8–14.9 | 38 | 2.502 | ARHGEF2 | Bos taurus Rho/Rac guanine nucleotide exchange factor (GEF) 2 |
| 3 | 14.8–14.9 | 38 | 2.502 | KIAA0907 | Bos taurus KH domain containing 4, pre-mRNA splicing factor |
| 3 | 14.8–14.9 | 38 | 2.502 | RXFP4 | Bos taurus relaxin family peptide/INSL5 receptor 4 |
| 3 | 14.8–14.9 | 38 | 2.502 | SSR2 | Bos taurus signal sequence receptor subunit 2 |
| 3 | 7.2–7.3 | 40 | 2.298 | NOS1AP | Bos taurus nitric oxide synthase 1 adaptor protein |
| 3 | 6.6–6.7 | 32 | 2.119 | DDR2 | Bos taurus discoidin domain receptor tyrosine kinase 2 |
| 3 | 6.6–6.7 | 32 | 2.119 | HSD17B7 | Bos taurus hydroxysteroid 17-beta dehydrogenase 7 |
| 5 | 69.2–69.3 | 16 | 2.332 | C5H12orf75 | Bos taurus chromosome 5 C12orf75 homolog |
| 5 | 37.3–37.4 | 16 | 2.261 | ADAMTS20 | Bos taurus ADAM metallopeptidase with thrombospondin type 1 motif 20 |
| 5 | 29.1–29.2 | 19 | 2.126 | METTL7A | Bos taurus methyltransferase like 7A |
| 5 | 28.3–28.4 | 38 | 1.982 | SCN8A | Bos taurus sodium voltage-gated channel alpha subunit 8 |
| 9 | 97.6–97.7 | 13 | 2.049 | AIRN | Bos taurus antisense IGF2R RNA noncoding |
| 9 | 97.6–97.7 | 13 | 2.049 | IGF2R | Bos taurus insulin-like growth factor 2 receptor |
| 10 | 11.5–11.6 | 42 | 2.415 | TXNDC16 | Bos taurus thioredoxin domain containing 16 |
| 10 | 11.4–11.5 | 28 | 2.05 | ERO1A | Bos taurus endoplasmic reticulum oxidoreductase 1 alpha |
| 10 | 11.4–11.5 | 28 | 2.05 | PSMC6 | Bos taurus proteasome 26S subunit, ATPase 6 |
| 10 | 10.4–10.5 | 22 | 1.985 | HOMER1 | Bos taurus homer scaffold protein 1 |
| 11 | 81.1–81.2 | 14 | 2.374 | VSNL1 | Bos taurus visinin like 1 |
| 13 | 60.7–60.8 | 14 | 1.945 | ANGPT4 | Bos taurus angiopoietin 4 |
| 13 | 60.7–60.8 | 14 | 1.945 | FAM110A | Bos taurus family with sequence similarity 110 member A |
| 17 | 63.5–63.6 | 39 | 2.169 | SLC8B1 | Bos taurus solute carrier family 8 member B1 |
| 22 | 49.2–49.3 | 13 | 1.958 | ALAS1 | Bos taurus 5′-aminolevulinate synthase 1 |
| 22 | 49.2–49.3 | 13 | 1.958 | POC1A | Bos taurus POC1 centriolar protein A |
| 22 | 49.2–49.3 | 13 | 1.958 | TLR9 | Bos taurus toll like receptor 9 |
| 22 | 49.2–49.3 | 13 | 1.958 | TWF2 | Bos taurus twinfilin actin binding protein 2 |
| 22 | 49.2–49.3 | 13 | 1.958 | WDR82 | Bos taurus WD repeat domain 82 |
| 26 | 45.9–46 | 30 | 2.162 | ADAM12 | Bos taurus ADAM metallopeptidase domain 12 |
| 28 | 33.5–33.6 | 26 | 2.277 | KCNMA1 | Bos taurus potassium calcium-activated channel subfamily M alpha 1 |
| 28 | 3.7–3.8 | 17 | 2.126 | ARV1 | Bos taurus ARV1 homolog, fatty acid homeostasis modulator |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Ma, H.; Xu, L.; Zhu, B.; Liu, Y.; Bordbar, F.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; et al. Genome-Wide Scan Identifies Selection Signatures in Chinese Wagyu Cattle Using a High-Density SNP Array. Animals 2019, 9, 296. https://doi.org/10.3390/ani9060296
Wang Z, Ma H, Xu L, Zhu B, Liu Y, Bordbar F, Chen Y, Zhang L, Gao X, Gao H, et al. Genome-Wide Scan Identifies Selection Signatures in Chinese Wagyu Cattle Using a High-Density SNP Array. Animals. 2019; 9(6):296. https://doi.org/10.3390/ani9060296
Chicago/Turabian StyleWang, Zezhao, Haoran Ma, Lei Xu, Bo Zhu, Ying Liu, Farhad Bordbar, Yan Chen, Lupei Zhang, Xue Gao, Huijiang Gao, and et al. 2019. "Genome-Wide Scan Identifies Selection Signatures in Chinese Wagyu Cattle Using a High-Density SNP Array" Animals 9, no. 6: 296. https://doi.org/10.3390/ani9060296