Reactive and Mixing Processes Governing Ammonium and Nitrate Coexistence in a Polluted Coastal Aquifer


 ,
,  ,
,  and
and
Abstract
1. Introduction
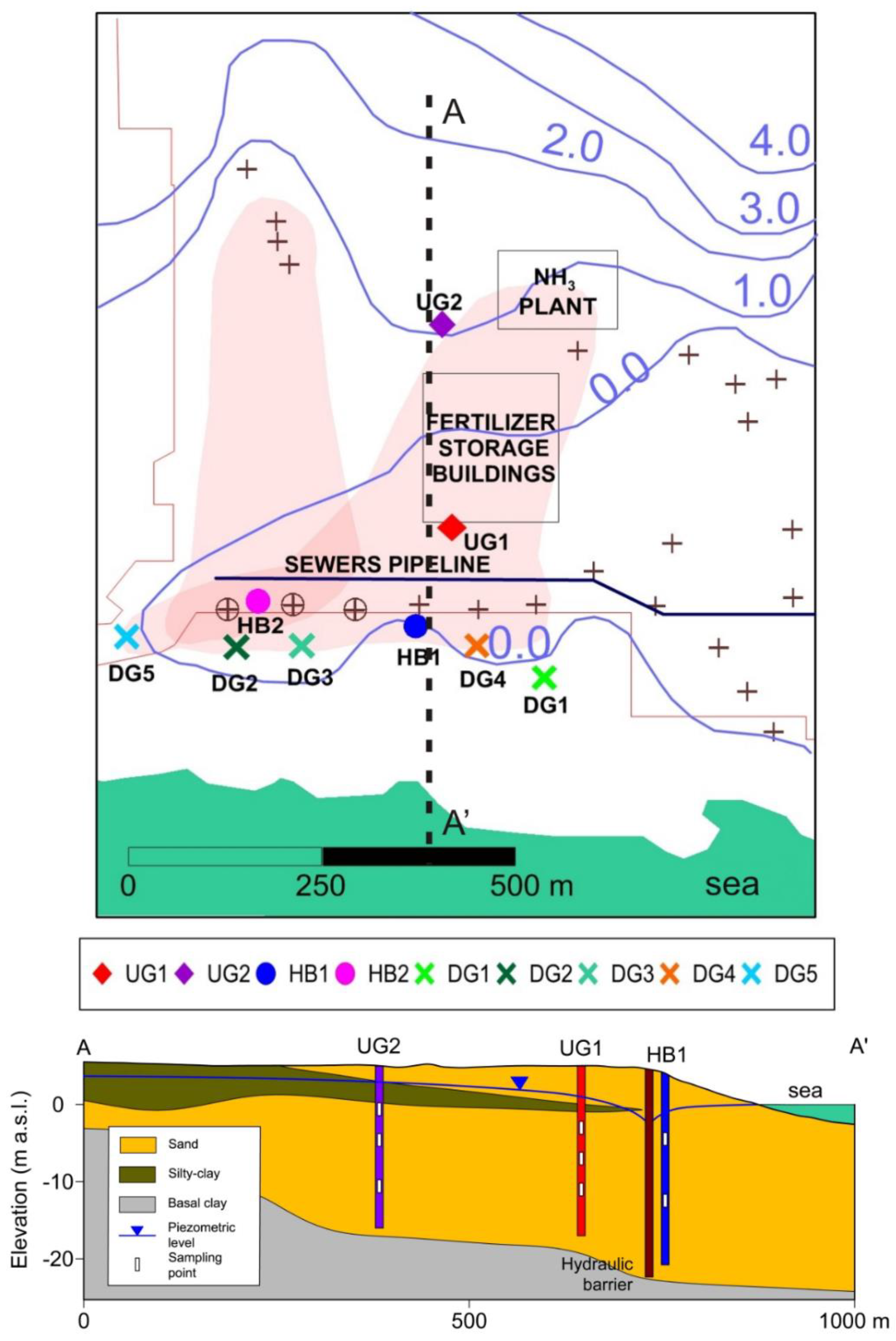
2. Geological and Hydrogeological Setting
3. Materials and Methods
3.1. Groundwater Sampling Technique and Chemical Analysis
3.2. Groundwater Isotopes Analyses
3.3. Nitrogen Isotope Analyses
3.4. Total Coliform Analysis
4. Results and Discussion
4.1. In Situ Measurements
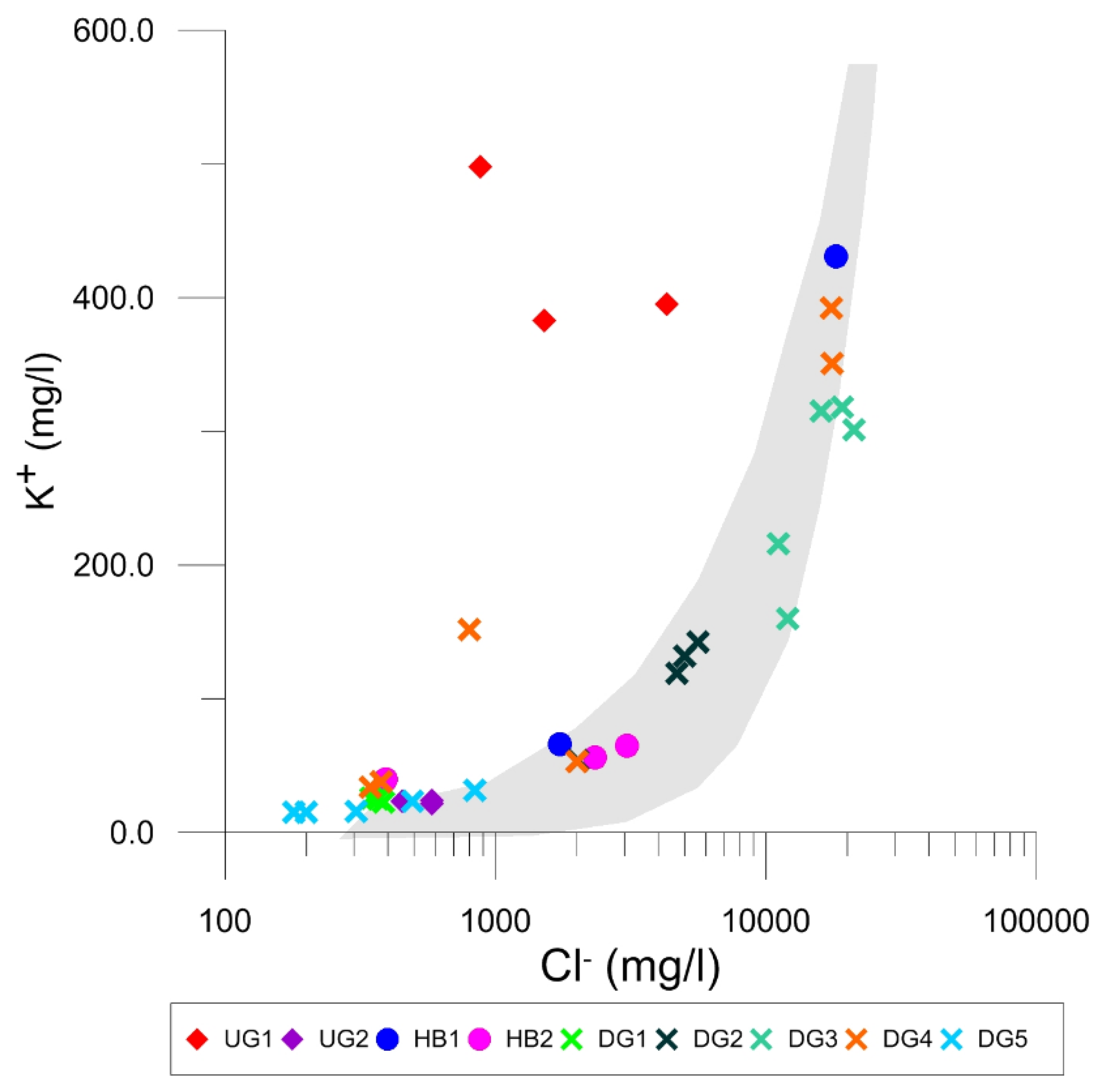
4.2. Cl− and K+ Concentration Patterns
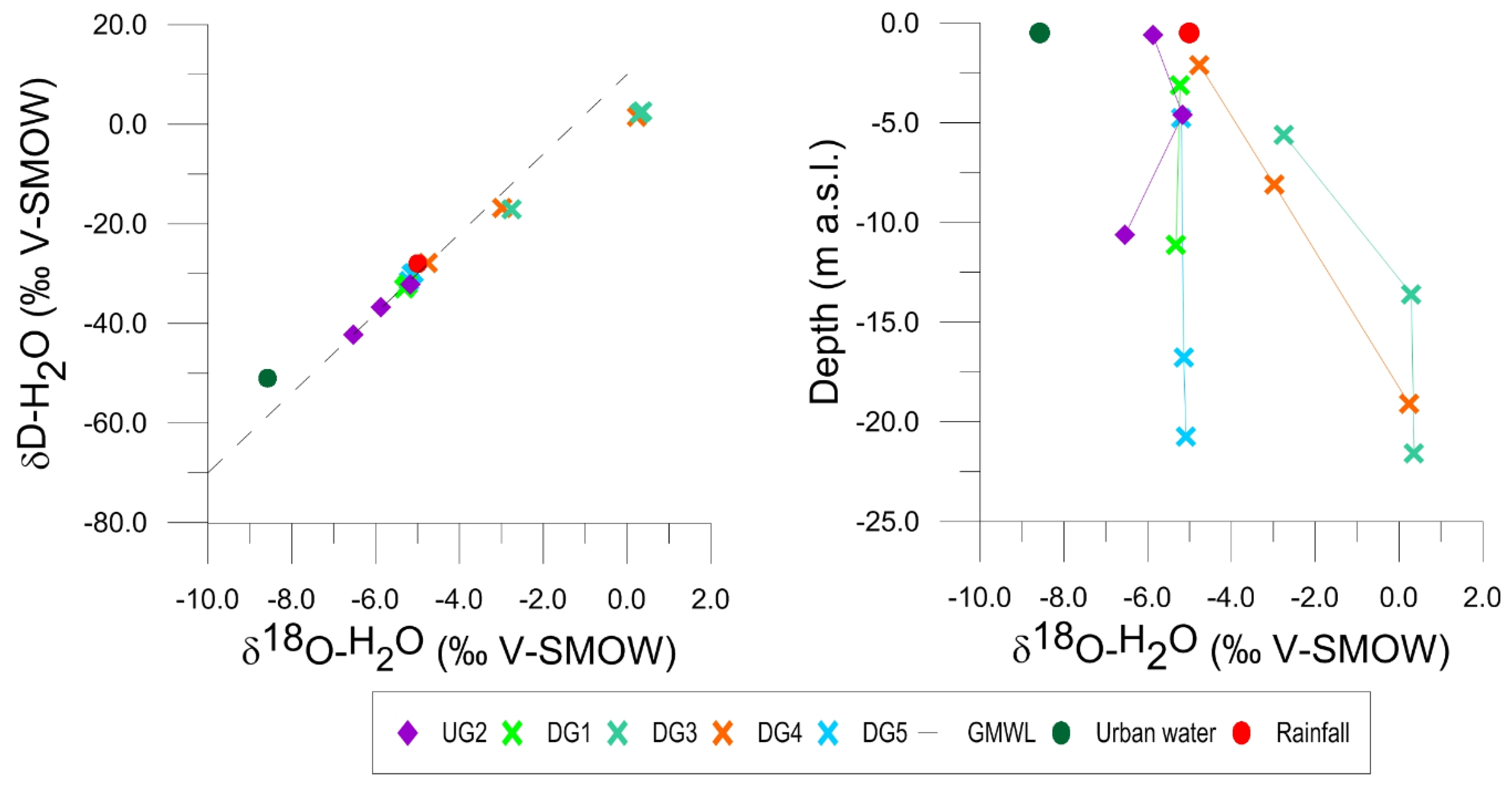
4.3. Stable Isotope Data
4.4. Nitrogen Pollution Sources and Degradation Processes Characterisation
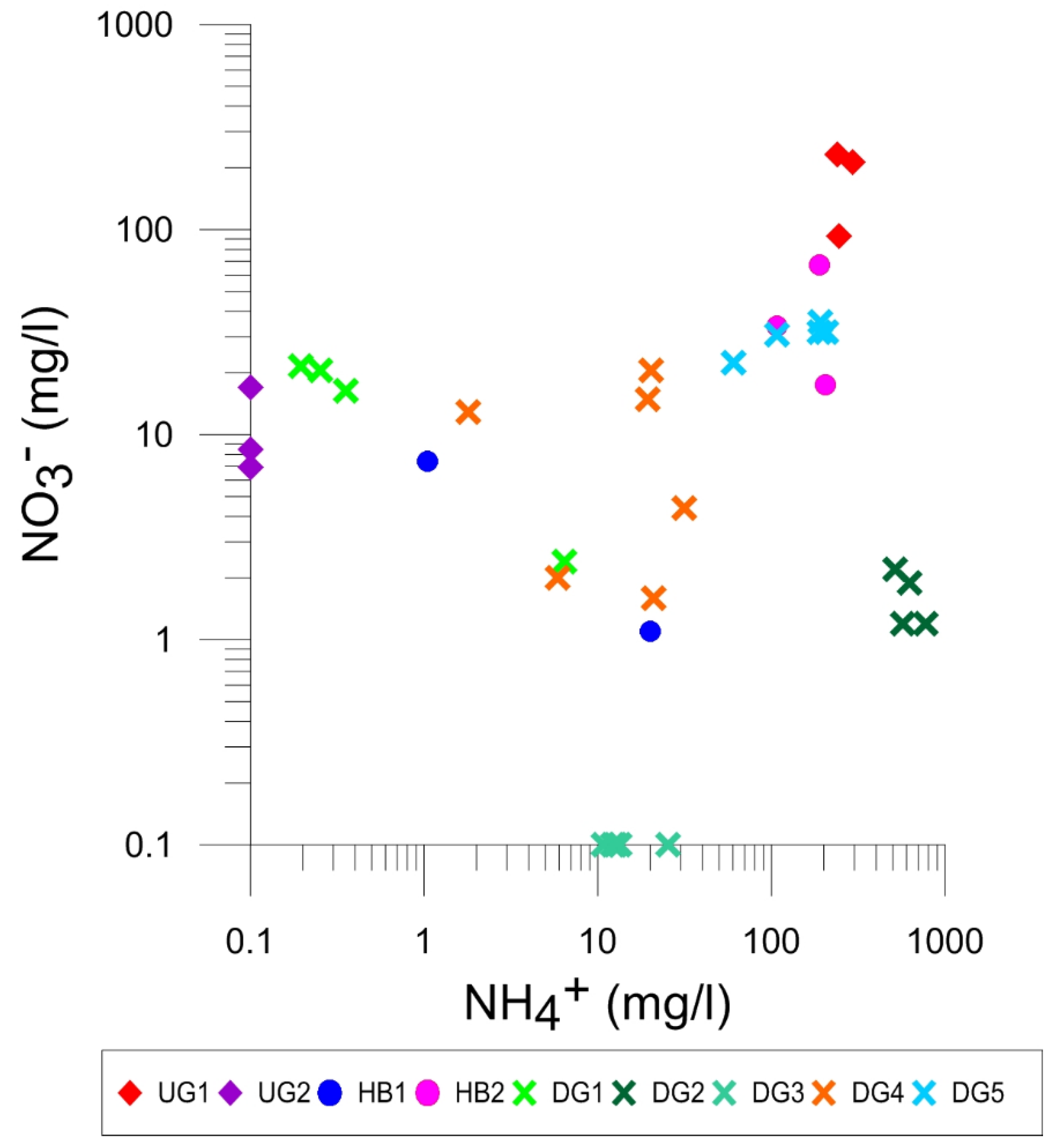
4.4.1. Concentration Patterns for Nitrogen Species and Coliforms
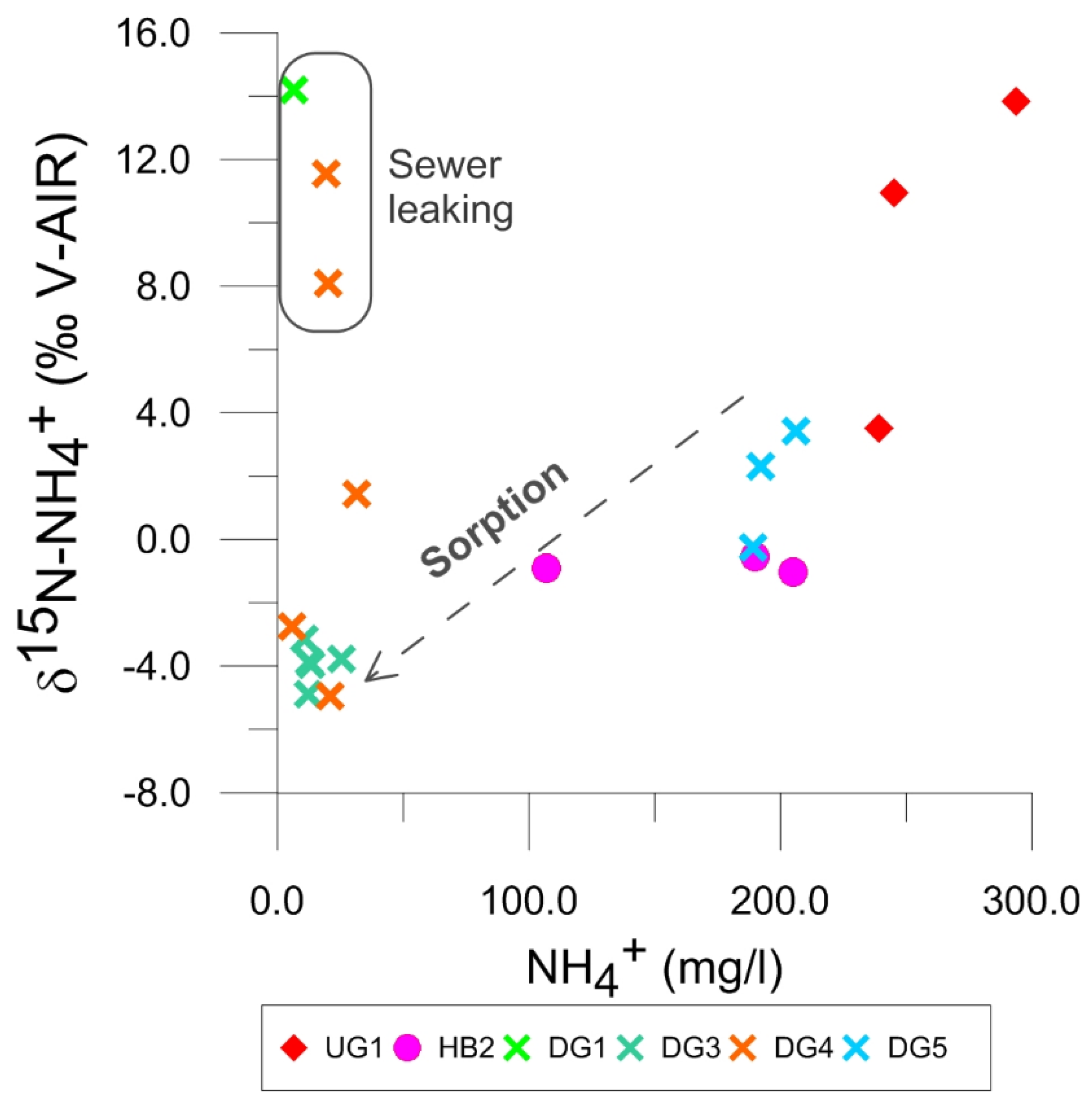
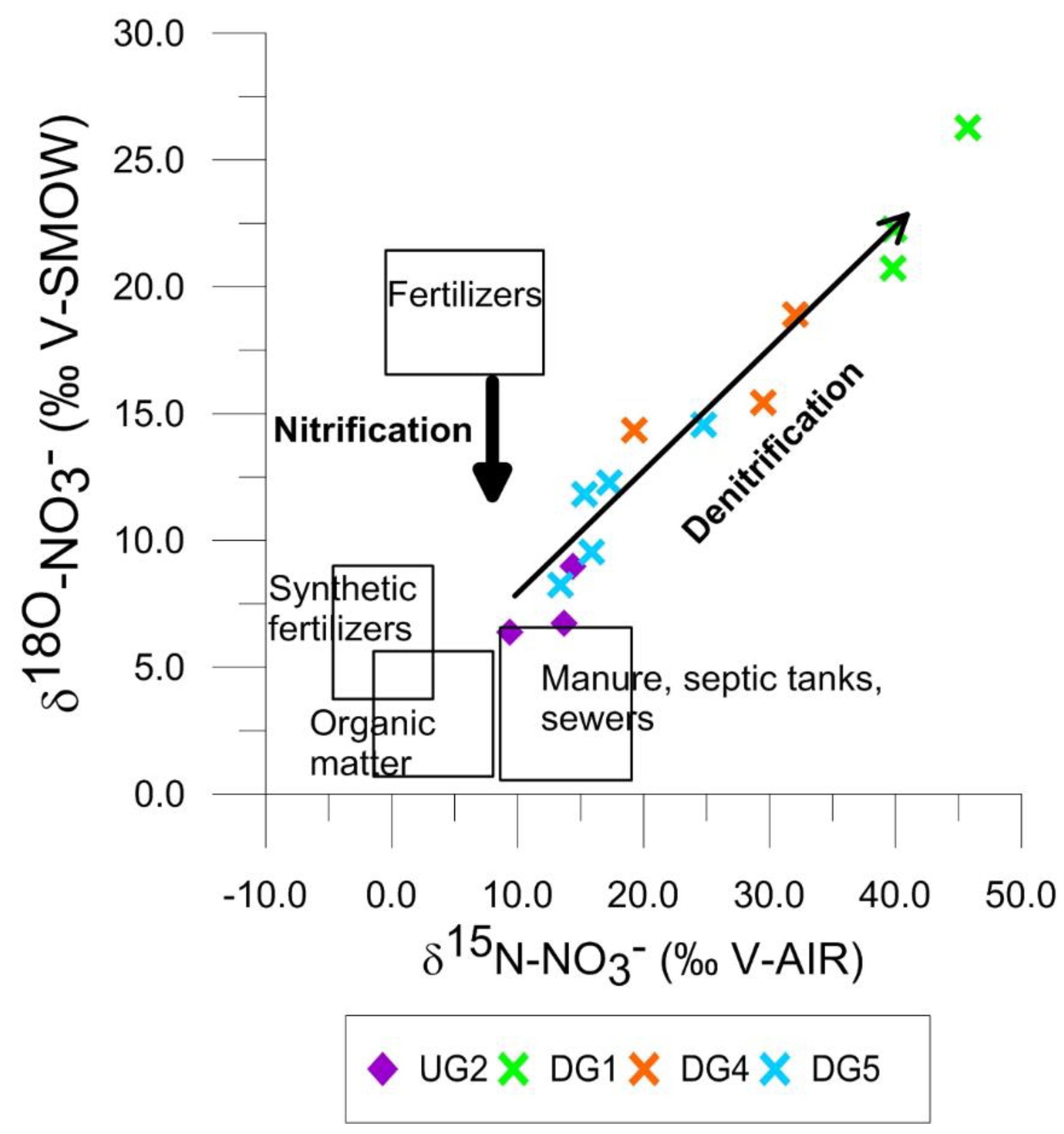
4.4.2. Isotope Data on NH4+ and NO3−
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Böhlke, J.K.; Smith, R.L.; Miller, D.N. Ammonium transport and reaction in contaminated groundwater: Application of isotope tracers and isotope fractionation studies. Water Resour. Res. 2006, 42, W05411. [Google Scholar] [CrossRef]
- Brauns, B.; Bjerg, P.L.; Song, X.; Jakobsen, R. Field scale interaction and nutrient exchange between surface water and shallow groundwater in the Baiyang Lake region, North China Plain. J. Environ. Sci. 2016, 45, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Zhang, Q.; Li, J.; Liu, Q. Tracing nitrate pollution sources and transformation in surface-and ground-waters using environmental isotopes. Sci. Total Environ. 2014, 490, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Best, A.; Arnaud, E.; Parker, B.; Aravena, R.; Dunfield, K. Effects of glacial sediment type and land use on nitrate patterns in groundwater. Groundw. Monit. Remediat. 2015, 35, 68–81. [Google Scholar] [CrossRef]
- Di Lorenzo, T.; Brilli, M.; Del Tosto, D.; Galassi, D.M.; Petitta, M. Nitrate source and fate at the catchment scale of the Vibrata River and aquifer (central Italy): An analysis by integrating component approaches and nitrogen isotopes. Environ. Earth Sci. 2012, 67, 2383–2398. [Google Scholar] [CrossRef]
- Petitta, M.; Fracchiolla, D.; Aravena, R.; Barbieri, M. Application of isotopic and geochemical tools for the evaluation of nitrogen cycling in an agricultural basin, the Fucino Plain, Central Italy. J. Hydrol. 2009, 372, 124–135. [Google Scholar] [CrossRef]
- Sebilo, M.; Mayer, B.; Nicolardot, B.; Pinay, G.; Mariotti, A. Long-term fate of nitrate fertilizer in agricultural soils. Proc. Natl. Acad. Sci. USA 2013, 110, 18185–18189. [Google Scholar] [CrossRef] [PubMed]
- Caschetto, M.; Robertson, W.; Petitta, M.; Aravena, R. Partial nitrification enhances natural attenuation of nitrogen in a septic system plume. Sci. Total Environ. 2018, 625, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Roehrdanz, P.R.; Feraud, M.; Lee, D.G.; Means, J.C.; Snyder, S.A.; Holden, P.A. Spatial models of sewer pipe leakage predict the occurrence of wastewater indicators in shallow urban groundwater. Environ. Sci. Technol. 2017, 51, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Toor, G.S. δ15N and δ18O reveal the sources of nitrate-nitrogen in urban residential stormwater runoff. Environ. Sci. Technol. 2016, 50, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.J.; Wang, Y.; Cherry, J.A.; Wang, X.; Zhi, B.; Du, H.; Wen, D. Abnormally high ammonium of natural origin in a coastal aquifer-aquitard system in the Pearl River Delta, China. Environ. Sci. Technol. 2010, 44, 7470–7475. [Google Scholar] [CrossRef] [PubMed]
- Mastrocicco, M.; Giambastiani, B.M.S.; Colombani, N. Ammonium occurrence in a salinized lowland coastal aquifer (Ferrara, Italy). Hydrol. Processes 2013, 27, 3495–3501. [Google Scholar] [CrossRef]
- Clark, I.; Timlin, R.; Bourbonnais, A.; Jones, K.; Lafleur, D.; Wickens, K. Origin and fate of industrial ammonium in anoxic ground water—15N evidence for anaerobic oxidation (anammox). Groundw. Monit. Remediat. 2008, 28, 73–82. [Google Scholar] [CrossRef]
- Colombani, N.; Mastrocicco, M.; Prommer, H.; Sbarbati, C.; Petitta, M. Fate of arsenic, phosphate and ammonium plumes in a coastal aquifer affected by saltwater intrusion. J. Contam. Hydrol. 2015, 179, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.; Heathwaite, L.; Binley, A.; Keenan, P. Nitrate concentration changes at the groundwater-surface water interface of a small Cumbrian river. Hydrol. Processes 2009, 23, 2195–2211. [Google Scholar] [CrossRef]
- Chen, S.; Ling, J.; Blancheton, J.P. Nitrification kinetics of biofilm as affected by water quality factors. Aquac. Eng. 2006, 34, 179–197. [Google Scholar] [CrossRef]
- Lansdown, K.; Heppell, C.M.; Trimmer, M.; Binley, A.; Heathwaite, A.L.; Byrne, P.; Zhang, H. The interplay between transport and reaction rates as controls on nitrate attenuation in permeable, streambed sediments. J. Geophys. Res. Biogeosci. 2015, 120, 1093–1109. [Google Scholar] [CrossRef]
- Snider, D.M.; Spoelstra, J.; Schiff, S.L.; Venkiteswaran, J.J. Stable oxygen isotope ratios of nitrate produced from nitrification: 18O-labeled water incubations of agricultural and temperate forest soils. Environ. Sci. Technol. 2010, 44, 5358–5364. [Google Scholar] [CrossRef] [PubMed]
- Sbarbati, C.; Colombani, N.; Mastrocicco, M.; Aravena, R.; Petitta, M. Performance of different assessment methods to evaluate contaminant sources and fate in a coastal aquifer. Environ. Sci. Pollut. Res. 2015, 22, 15536–15548. [Google Scholar] [CrossRef] [PubMed]
- Philips, S.; Laanbroek, H.J.; Verstraete, W. Origin, causes and effects of increased nitrite concentrations in aquatic environments. Rev. Environ. Sci. Biotechnol. 2002, 1, 115–141. [Google Scholar] [CrossRef]
- Caschetto, M.; Colombani, N.; Mastrocicco, M.; Petitta, M.; Aravena, R. Nitrogen and sulphur cycling in the saline coastal aquifer of Ferrara, Italy. A multi-isotope approach. Appl. Geochem. 2017, 76, 88–98. [Google Scholar] [CrossRef]
- Ding, J.; Xi, B.; Xu, Q.; Su, J.; Huo, S.; Liu, H.; Yu, Y.; Zhang, Y. Assessment of the sources and transformations of nitrogen in a plain river network region using a stable isotope approach. J. Environ. Sci. 2015, 30, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yang, Y.; Lian, X.; Jiang, Y.; Xi, B.; Peng, X.; Yan, K. Identification of nitrate sources in groundwater using a stable isotope and 3DEEM in a landfill in Northeast China. Sci. Total Environ. 2016, 563, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Martínez, D.; Moschione, E.; Bocanegra, E.; Galli, M.G.; Aravena, R. Distribution and origin of nitrate in groundwater in an urban and suburban aquifer in Mar del Plata, Argentina. Environ. Earth Sci. 2014, 72, 1877–1886. [Google Scholar] [CrossRef]
- Minet, E.P.; Goodhue, R.; Meier-Augenstein, W.; Kalin, R.M.; Fenton, O.; Richards, K.G.; Coxon, C.E. Combining stable isotopes with contamination indicators: A method for improved investigation of nitrate sources and dynamics in aquifers with mixed nitrogen inputs. Water Res. 2017, 124, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Puig, R.; Soler, A.; Widory, D.; Mas-Pla, J.; Domènech, C.; Otero, N. Characterizing sources and natural attenuation of nitrate contamination in the Baix Ter aquifer system (NE Spain) using a multi-isotope approach. Sci. Total Environ. 2017, 580, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Wells, N.S.; Hakoun, V.; Brouyère, S.; Knöller, K. Multi-species measurements of nitrogen isotopic composition reveal the spatial constraints and biological drivers of ammonium attenuation across a highly contaminated groundwater system. Water Res. 2016, 98, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Mastrocicco, M.; Colombani, N.; Sbarbati, C.; Petitta, M. Assessing the effect of saltwater intrusion on petroleum hydrocarbons plumes via numerical modelling. Water Air Soil Pollut. 2012, 223, 4417–4427. [Google Scholar] [CrossRef]
- Mastrocicco, M.; Sbarbati, C.; Colombani, N.; Petitta, M. Efficiency verification of a horizontal flow barrier via flowmeter tests and multilevel sampling. Hydrol. Processes 2013, 27, 2414–2421. [Google Scholar] [CrossRef]
- Epstein, S.; Mayeda, T. Variation of O-18 content of waters from natural sources. Geochim. Cosmichim. Acta 1953, 4, 213–224. [Google Scholar] [CrossRef]
- Longinelli, A.; Selmo, E. Isotopic composition of precipitation in Italy: A first overall map. J. Hydrol. 2003, 270, 75–88. [Google Scholar] [CrossRef]
- Coleman, M.L.; Sheppard, T.J.; Durham, J.J.; Rouse, J.E.; Moore, G.R. Reduction of water with zinc for hydrogen isotope analyses. Anal. Chem. 1982, 54, 993–995. [Google Scholar] [CrossRef]
- Brooks, P.D.; Stark, J.M.; McInteer, B.B.; Preston, T. Diffusion method to prepare soil extracts for automated nitrogen-15 analysis. Soil Sci. Soc. Am. J. 1989, 53, 1707–1711. [Google Scholar] [CrossRef]
- Sørensen, P.; Jensen, E.S. Sequential diffusion of ammonium and nitrate from soil extracts to a polytetrafluoroethylene trap for 15N determination. Anal. Chim. Acta 1991, 252, 201–203. [Google Scholar] [CrossRef]
- Silva, S.R.; Kendall, C.; Wilkison, D.H.; Ziegler, A.C.; Chang, C.C.Y.; Avanzino, R.J. A new method for collection of nitrate from fresh water and the analysis of nitrogen and oxygen isotope ratios. J. Hydrol. 2009, 228, 22–36. [Google Scholar] [CrossRef]
- Casciotti, K.L.; Sigman, D.M.; Hastings, M.; Böhlke, J.K.; Hilkert, A. Measurement of the oxygen isotopic composition of nitrate in seawater and freshwater using the denitrifier method. Anal. Chem. 2002, 74, 4905–4912. [Google Scholar] [CrossRef] [PubMed]
- Rock, L.; Ellert, B.H. Nitrogen-15 and oxygen-18 natural abundance of potassium chloride extractable soil nitrate using the denitrifier method. Soil Sci. Soc. Am. J. 2007, 71, 355–361. [Google Scholar] [CrossRef]
- Sigman, D.M.; Casciotti, K.L.; Andreani, M.; Barford, C.; Galanter, M.; Böhlke, J.K. A bacterial method for the nitrogen isotopic analysis of nitrate in seawater and freshwater. Anal. Chem. 2002, 73, 4145–4153. [Google Scholar] [CrossRef]
- Hurley, M.A.; Roscoe, M.E. Automated statistical-analysis of microbial enumeration by dilution series. J. Appl. Microbiol. 1983, 55, 159–164. [Google Scholar] [CrossRef]
- Aravena, R.; Mayer, B. Isotopes and Processes in the Nitrogen and Sulfur Cycles. In Environmental Isotopes in Biodegradation and Bioremediation; CRC Press/Lewis: Boca Raton, FL, USA, 2010; pp. 203–246. ISBN 9781566706612. [Google Scholar]
- Robertson, W.D.; Moore, T.A.; Spoelstra, J.; Li, L.; Elgood, R.J.; Clark, I.D.; Schiff, S.L.; Aravena, R.; Neufeld, J.D. Natural attenuation of septic system nitrogen by anammox. Groundwater 2012, 50, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Aravena, R.; Robertson, W. The use of multiple isotope tracers to evaluate denitrification in groundwater: A case study in a large septic system plume. Groundwater 1998, 36, 975–982. [Google Scholar] [CrossRef]
- Clark, I.; Fritz, P. Environmental Isotopes in Hydrogeology; CRC Press/Lewis: Boca Raton, FL, USA, 1997; ISBN 1566702496. [Google Scholar]
- Mariotti, A.; Germon, J.C.; Hubert, P.; Kaiser, P.; Letolle, R.; Tardieux, A.; Tardieux, P. Experimental determination of nitrogen kinetic isotope fractionation: Some principles; Illustration for the denitrification and nitrification processes. Plant Soil 1981, 62, 413–430. [Google Scholar] [CrossRef]
- Karamanos, R.E.; Rennie, D.A. Nitrogen isotope fractionation during ammonium exchange reactions with soil clay. Canad. J. Soil Sci. 1978, 58, 53–60. [Google Scholar] [CrossRef]
- Sbarbati, C. Use of an Integrated Methodological Approach to Assess Contaminant Fate and Transport in a Coastal Aquifer. Ph.D. Thesis, Sapienza University of Rome, Rome, Italy, 2013. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples ID | Elevation (m a.s.l.) | EC (μS/cm) | Eh (mV) | Cl (mg/L) | K (mg/L) | δ18O–H2O | δD–H2O | NH4 (mg/L) | NO3 (mg/L) | NO2 (μg/L) | δ15N–NH4 | δ15N–NO3 | δ18O–NO3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Detection limit | 0.1 | 0.4 | 0.1 | 0.1 | 10 | ||||||||
| UG1a | −3.02 | 7800 | −24 | 880.0 | 498.0 | - | - | 293.8 | 214.0 | 52.12 | 13.8 | - | - |
| UG1b | −7.02 | 10,200 | −60 | 1510.0 | 383.0 | - | - | 245.0 | 93.0 | 29.00 | 10.9 | - | - |
| UG1c | −11.02 | 27,400 | −137 | 4300.0 | 395.0 | - | - | 239.0 | 233.0 | >10 | 3.5 | - | - |
| UG2a | −0.61 | 168 | 3 | 452.0 | 23.6 | −5.9 | −36.8 | >0.1 | 17.0 | >10 | - | 9.4 | 6.4 |
| UG2b | −4.61 | 159 | 2 | 580.0 | 23.8 | −5.2 | −32.2 | >0.1 | 8.5 | >10 | - | 13.7 | 6.7 |
| UG2c | −10.61 | 313 | 2 | 580.0 | 21.3 | −6.5 | −42.3 | >0.1 | 6.9 | >10 | - | 14.4 | 8.9 |
| HB1a | −4.47 | 35,887 | −180 | 1730.0 | 66.0 | - | - | 1.0 | 7.4 | 22.20 | - | - | - |
| HB1b | −12.47 | 39,516 | −185 | 18,200.0 | 431.0 | - | - | 20.1 | 1.1 | 25.00 | - | - | - |
| HB2a | −5.18 | 43,800 | −61 | 393.0 | 39.5 | - | - | 107.0 | 33.7 | 150.01 | −0.9 | - | - |
| HB2b | −9.18 | 45,900 | −74 | 2340.0 | 56.0 | - | - | 190.0 | 67.3 | 134.00 | −0.6 | - | - |
| HB2c | −13.18 | 46,100 | −95 | 3060.0 | 65.0 | - | - | 205.0 | 17.5 | >10 | −1.0 | - | - |
| DG1a | −3.12 | 2245 | 276 | 386.0 | 23.0 | −5.2 | −32.2 | 6.5 | 2.4 | 25.10 | 14.9 | - | - |
| DG1b | −7.12 | 2378 | 241 | 347.0 | 25.9 | - | - | 0.4 | 16.4 | >10 | - | 45.8 | 26.3 |
| DG1c | −11.12 | 2368 | 226 | 369.0 | 24.9 | −5.3 | −33 | 0.3 | 20.6 | >10 | - | 39.9 | 22.3 |
| DG1d | −19.12 | 2369 | 208 | 368.0 | 25.5 | - | - | 0.2 | 21.6 | >10 | - | 39.8 | 20.7 |
| DG2a | −7.52 | 36,523 | −68 | 2020.0 | 53.8 | - | - | 516.0 | 2.2 | 23.04 | - | - | - |
| DG2b | −11.52 | 37,412 | −83 | 4680.0 | 119.0 | - | - | 569.0 | 1.2 | 17.30 | - | - | - |
| DG2c | −15.52 | 42,631 | −96 | 5000.0 | 131.9 | - | - | 627.0 | 1.9 | 15.00 | - | - | - |
| DG2d | −19.52 | 43,203 | −101 | 5600.0 | 142.4 | - | - | 769.0 | 1.2 | <10 | - | - | - |
| DG3a | −5.60 | 43,388 | −73 | 12,000.0 | 160.0 | −2.8 | −17.1 | 12.6 | >0.1 | 53.80 | −3.9 | - | - |
| DG3b | −9.60 | 50,832 | −96 | 11,100.0 | 216.0 | - | - | 25.4 | >0.1 | >10 | −3.8 | - | - |
| DG3c | −13.60 | 50,822 | −101 | 21,200.0 | 301.0 | 0.3 | 2.2 | 13.3 | >0.1 | >10 | −3.9 | - | - |
| DG3d | −17.60 | 50,864 | −104 | 19,200.0 | 318.0 | - | - | 12.2 | >0.1 | >10 | −4.9 | - | - |
| DG3e | −21.60 | 49,653 | −93 | 16,000.0 | 315.0 | 0.4 | 2.6 | 10.8 | >0.1 | >10 | −3.2 | - | - |
| DG4a | −2.10 | 2441 | 123 | 344.0 | 33.9 | −4.8 | −27.8 | 19.3 | 14.9 | 3040.00 | 11.6 | 32.1 | 18.9 |
| DG4b | −5.10 | 2506 | 125 | 377.0 | 37.9 | - | - | 20.3 | 20.5 | 1520.00 | 8.1 | 29.5 | 15.4 |
| DG4c | −8.10 | 2504 | 129 | 2000.0 | 53.0 | −2.9 | −16.9 | 1.8 | 12.9 | 1070.00 | − | 19.2 | 14.4 |
| DG4d | −11.10 | 10,614 | 64 | 17,600.0 | 351.0 | - | - | 20.9 | 1.6 | >10 | −4.9 | - | - |
| DG4e | −15.10 | 48,052 | −55 | 800.0 | 152.0 | - | - | 31.5 | 4.4 | >10 | 1.4 | - | - |
| DG4f | −19.10 | 50,375 | −70 | 17,500.0 | 392.0 | 0.2 | 1.5 | 5.9 | 2.0 | >10 | −2.8 | - | - |
| DG5a | −4.76 | 4074 | 301 | 180.0 | 15.0 | −5.2 | −31.5 | 189.0 | 31.7 | 123.30 | −0.2 | 15.9 | 9.5 |
| DG5b | −8.76 | 4572 | 299 | 306.0 | 15.5 | - | - | 206.0 | 31.5 | 144.00 | 3.4 | 17.3 | 12.3 |
| DG5c | −12.76 | 5132 | 263 | 199.0 | 15.3 | - | - | 192.0 | 35.4 | 142.21 | 2.3 | 13.4 | 8.2 |
| DG5d | −16.76 | 28,115 | 68 | 840.0 | 31.5 | −5.1 | −30.2 | 107.9 | 30.7 | 135.07 | −1.0 | 15.3 | 11.8 |
| DG5e | −20.76 | 36,629 | −25 | 493.0 | 23.4 | −5.1 | −29.9 | 60.4 | 22.4 | 149.00 | −0.9 | 24.8 | 14.6 |
| Rainfall | - | - | - | - | - | −5.0 | −28.0 | - | - | - | - | - | - |
| Urban water | - | - | - | - | - | −8.6 | −51.0 | - | - | - | - | - | - |
| Sample Name | Value (MPN/100 mL) |
|---|---|
| Upgradient monitoring well | Below detection limits |
| Sewer pipeline | <100,000 |
| Monitoring well 1 at the hydraulic barrier | 145 |
| Monitoring well 2 at the hydraulic barrier | 300 |
| Monitoring well 3 at the hydraulic barrier | 8719 |
| Pumping well 1 of the hydraulic barrier | Below detection limits |
| Pumping well 2 of the hydraulic barrier | Below detection limits |
| Pumping well 3 of the hydraulic barrier | Below detection limits |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sbarbati, C.; Colombani, N.; Mastrocicco, M.; Petitta, M.; Aravena, R. Reactive and Mixing Processes Governing Ammonium and Nitrate Coexistence in a Polluted Coastal Aquifer. Geosciences 2018, 8, 210. https://doi.org/10.3390/geosciences8060210
Sbarbati C, Colombani N, Mastrocicco M, Petitta M, Aravena R. Reactive and Mixing Processes Governing Ammonium and Nitrate Coexistence in a Polluted Coastal Aquifer. Geosciences. 2018; 8(6):210. https://doi.org/10.3390/geosciences8060210
Chicago/Turabian StyleSbarbati, Chiara, Nicolò Colombani, Micòl Mastrocicco, Marco Petitta, and Ramon Aravena. 2018. "Reactive and Mixing Processes Governing Ammonium and Nitrate Coexistence in a Polluted Coastal Aquifer" Geosciences 8, no. 6: 210. https://doi.org/10.3390/geosciences8060210
APA StyleSbarbati, C., Colombani, N., Mastrocicco, M., Petitta, M., & Aravena, R. (2018). Reactive and Mixing Processes Governing Ammonium and Nitrate Coexistence in a Polluted Coastal Aquifer. Geosciences, 8(6), 210. https://doi.org/10.3390/geosciences8060210