Fifteen Years of Radionuclide Research at the KIT Synchrotron Source in the Context of the Nuclear Waste Disposal Safety Case

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Performing Experiments with Radionuclides at the KIT Synchrotron Source
3. Highlights of Past Research Projects
4. Examples for Ongoing Research Projects
4.1. Aquatic Chemistry of Neptunium under Hyperalkaline and Oxidizing Conditions
4.1.1. Experimental
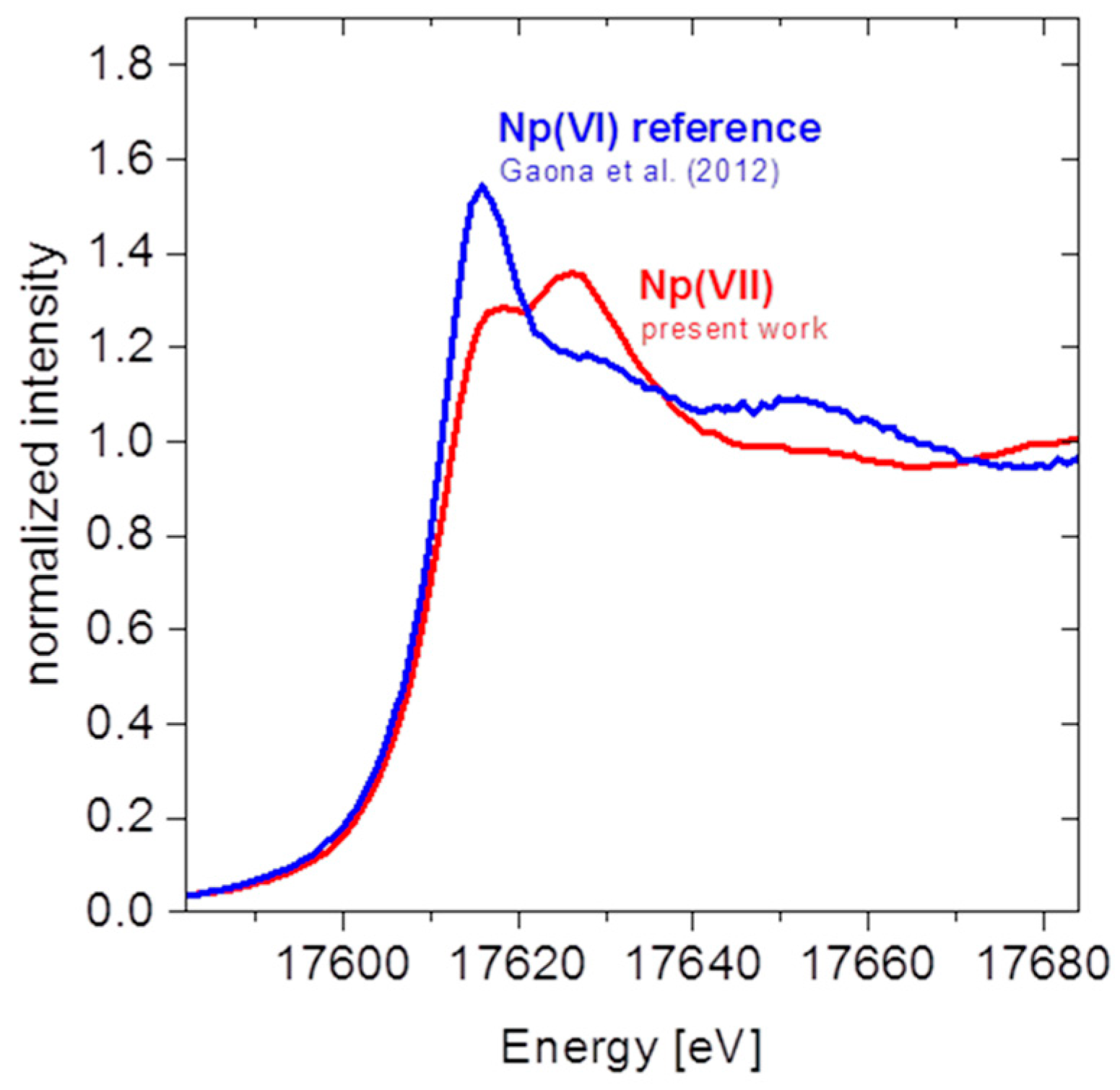
4.1.2. Results and Discussion
log K°VIaq-VIIaq = log [NpO4(OH)23−] + log γ {NpO4(OH)23−} − pe + 2 log aw − log [NpO2(OH)42−]
− log γ {NpO2(OH)42−} − 2 log [OH−] − 2 log γ {OH−}
log K°VIs-VIIaq = log [NpO4(OH)23−] + log γ {NpO4(OH)23−} + log [Na+] + log γ {Na+} − pe + 0.5 log aw
− 3 log [OH−] − 3 log γ {OH−}
4.2. Speciation of Actinides in a Spent Nuclear Fuel (Snf) Bulk Fragment
4.2.1. Experimental
4.2.2. Results and Discussion
4.3. High Resolution Xanes Study of the Influence Of Fe(II) on the Redox State of U Sorbed on Magnetite
4.3.1. Experimental
4.3.2. Results and Discussion
5. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ewing, R.C. Long-term storage of spent nuclear fuel. Nat. Mater. 2015, 14, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Ewing, R.C.; Whittleston, R.A.; Yardley, B.W.D. Geological disposal of nuclear waste: A primer. Elements 2016, 12, 234–239. [Google Scholar] [CrossRef]
- Zimina, A.; Dardenne, K.; Denecke, M.A.; Grunwald, J.-D.; Huttel, E.; Lichtenberg, H.; Mangold, S.; Prüßmann, T.; Rothe, J.; Steininger, R.; et al. The CAT-ACT Beamline at ANKA: A new high energy X-ray spectroscopy facility for CATalysis and ACTinide research. J. Phys. Conf. Ser. 2016, 712, 012019. [Google Scholar] [CrossRef]
- The KIT Institute for Beam Physics and Technology (IBPT). Available online: www.ibpt.kit.edu/ (accessed on 11 December 2018).
- Denecke, M.A. X-ray Spectroscopy in Studies of the Nuclear Fuel Cycle. In X-ray Absorption and X-ray Emission Spectroscopy: Theory and Applications; van Bokhoven, J.A., Lamberti, C., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2016; ISBN 978-1-118-84428-1. [Google Scholar]
- Rothe, J.; Butorin, S.; Dardenne, K.; Denecke, M.A.; Kienzler, B.; Löble, M.; Metz, V.; Seibert, A.; Steppert, M.; Vitova, T.; et al. The INE-Beamline for actinide science at ANKA. Rev. Sci. Instrum. 2012, 83, 043105. [Google Scholar] [CrossRef] [PubMed]
- Zimina, A.; Dardenne, K.; Denecke, M.A.; Doronkin, D.E.; Huttel, E.; Lichtenberg, H.; Mangold, S.; Prüßmann, T.; Rothe, J.; Spangenberg, T.; et al. CAT-ACT—A new highly versatile X-ray spectroscopy beamline for catalysis and radionuclide science at the KIT synchrotron light facility ANKA. Rev. Sci. Instrum. 2017, 88, 113113. [Google Scholar] [CrossRef] [PubMed]
- Transnational Access to Large Infrastructure for a Safe Management of ActiNide (TALISMAN). Available online: www.actinet-i3.eu/ (accessed on 11 December 2018).
- Hennig, C.; Tutschku, J.; Rossberg, A.; Bernhard, G.; Scheinost, A.C. Comparative EXAFS Investigation of Uranium(VI) and -(IV) Aquo Chloro Complexes in Solution Using a Newly Developed Spectroelectrochemical Cell. Inorg. Chem. 2005, 44, 6655–6661. [Google Scholar] [CrossRef] [PubMed]
- Gaona, X.; Tits, J.; Dardenne, K.; Liu, X.; Rothe, J.; Denecke, M.A.; Wieland, E.; Altmaier, M. Spectroscopic investigations of Np(V/VI) redox speciation in hyperalkaline TMA-(OH,Cl) solutions. Radiochim. Acta 2012, 100, 759–770. [Google Scholar] [CrossRef]
- Skerencak-Frech, A.; Fröhlich, D.R.; Rothe, J.; Dardenne, K.; Panak, P.J. Combined Time-Resolved Laser Fluorescence Spectroscopy and Extended X-ray Absorption Fine Structure Spectroscopy Study on the Complexation of Trivalent Actinides with Chloride at T = 25–200 degrees C. Inorg. Chem. 2014, 53, 1062–1069. [Google Scholar] [CrossRef]
- Prieur, D.; Lebreton, F.; Caisso, M.; Martin, P.M.; Scheinost, A.C.; Delahaye, T.; Manara, D. Melting behaviour of americium-doped uranium dioxide. J. Chem. Thermodyn. 2016, 97, 244–252. [Google Scholar] [CrossRef]
- Lebreton, F.; Horlait, D.; Delahaye, T.; Blanchart, P. Fabrication and characterization of U1−xAmxO2±δ compounds with high americium contents (x = 0.3, 0.4 and 0.5). J. Nucl. Mater. 2013, 439, 99–102. [Google Scholar] [CrossRef]
- Caisso, M.; Picart, S.; Belin, R.C.; Lebreton, F.; Martin, P.M.; Dardenne, K.; Rothe, J.; Neuville, D.R.; Delahaye, T.; Ayral, A. In situ characterization of uranium and americium oxide solid solution formation for CRMP process: First combination of in situ XRD and XANES measurements. Dalton Trans. 2015, 44, 6391–6399. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, J.; Grünewald, W.; Roth, G.; Schmitz, F.-J.; Tobie, W.; Weishaupt, M. Successful Hot Operation of the German Vitrification Plant VEK—Results and Experiences. In Proceedings of the WM 2012-Conference, Phoenix, AZ, USA, 26 February–1 March 2012; No. 11277. pp. 1–7. [Google Scholar]
- Dardenne, K.; González-Robles, E.; Rothe, J.; Müller, N.; Christill, G.; Lemmer, D.; Praetorius, R.; Kienzler, B.; Metz, V.; Roth, G.; et al. XAS and XRF investigation of a genuine HAWC glass fragment obtained from the Karlsruhe vitrification plant (VEK). J. Nucl. Mater. 2015, 460, 209–215. [Google Scholar] [CrossRef]
- Grambow, B. Nuclear waste glasses—How durable. Elements 2006, 2, 357–364. [Google Scholar] [CrossRef]
- Rothe, J.; Walther, C.; Denecke, M.A.; Fanghänel, T. XAFS and LIBD Investigation of the Formation and Structure of Colloidal Pu(IV) Hydrolysis Products. Inorg. Chem. 2004, 43, 4708–4718. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.; Rothe, J.; Brendebach, B.; Fuss, M.; Altmaier, M.; Marquardt, C.M.; Büchner, S.; Cho, H.-R.; Yun, J.-I.; Seibert, A. New insights in the formation processes of Pu(IV) colloids. Radiochim. Acta 2009, 97, 199–207. [Google Scholar] [CrossRef]
- Kersting, A.B. Plutonium Transport in the Environment. Inorg. Chem. 2013, 52, 3533–3546. [Google Scholar] [CrossRef] [PubMed]
- Tasi, A.; Gaona, X.; Fellhauer, D.; Böttle, M.; Rothe, J.; Dardenne, K.; Schild, D.; Grivé, M.; Colàs, E.; Bruno, J.; et al. Redox behavior and solubility of plutonium under alkaline, reducing conditions. Radiochim. Acta 2018, 106, 259–279. [Google Scholar] [CrossRef]
- Tasi, A.; Gaona, X.; Fellhauer, D.; Böttle, M.; Rothe, J.; Dardenne, K.; Polly, R.; Grivé, M.; Colàs, E.; Bruno, J.; et al. Thermodynamic description of the plutonium–α–d–isosaccharinic acid system ii: Formation of quaternary Ca(II)–Pu(IV)–OH–ISA complexes. Appl. Geochem. 2018, 98, 351–366. [Google Scholar] [CrossRef]
- Clark, D.L.; Geeson, D.A.; Hanrahan, R.J., Jr. (Eds.) Plutonium Handbook, 2nd ed.; American Nuclear Society: La Grange Park, IL, USA, 2018. [Google Scholar]
- Guillaumont, R.; Fanghänel, T.; Fuger, J.; Grenthe, I.; Neck, V.; Palmer, D.A.; Rand, M.H. Update on the Chemical Thermodynamics of U, Np, Pu, Am and Tc; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Morss, L.R.; Edelstein, N.M.; Fuger, J.; Katz, J.J. (Eds.) The Chemistry of the Actinide and Transactinide Elements; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Altmaier, M.; Gaona, X.; Fanghänel, T. Recent Advances in Aqueous Actinide Chemistry and Thermodynamics. Chem. Rev. 2013, 113, 901–943. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.W.; Blaudeau, J.P.; Sullivan, J.C.; Antonio, M.R.; Bursten, B.; Soderholm, L. The Coordination Geometry of Np(VII) in Alkaline Solution. J. Am. Chem. Soc. 2001, 123, 4346–4347. [Google Scholar] [CrossRef] [PubMed]
- Bolvin, H.; Wahlgren, U.; Moll, H.; Reich, T.; Geipel, G.; Fanghänel, T.; Grenthe, I. On the structure of Np(VI) and Np(VII) species in alkaline solution studied by EXAFS and quantum chemical methods. J. Phys. Chem. A 2001, 105, 11441–11445. [Google Scholar] [CrossRef]
- Clark, D.L.; Palmer, P.D.; Tait, C.D.; Webster Keogh, D.W.; Conradson, S.D.; Donohoe, R.J. Actinide Research Quarterly 2004, 1st Quarter. Available online: https://www.lanl.gov/orgs/nmt/nmtdo/AQarchive/04spring/ (accessed on 11 December 2018).
- Ikeda-Ohno, A.; Tsushima, S.; Takao, K.; Rossberg, A.; Funke, H.; Scheinost, A.C.; Bernhard, G.; Yaita, T.; Hennig, C. Neptunium Carbonato Complexes in Aqueous Solution: An Electrochemical, Spectroscopic, and Quantum Chemical Study. Inorg. Chem. 2009, 48, 11779–11787. [Google Scholar] [CrossRef]
- Lozano, J.M.; Clark, D.L.; Conradson, S.D.; Den Auwer, C.; Fillaux, C.; Guilaumont, D.; Webster Keogh, D.; Mustre De Leon, J.; Palmer, P.D.; Simoni, E. Influence of the local atomic structure in the X-ray absorption near edge spectroscopy of neptunium oxo ions. Phys. Chem. Chem. Phys. 2009, 11, 10396–10402. [Google Scholar] [CrossRef] [PubMed]
- Wren, J.E.C.; Schreckenbach, G. Neptunium(VII) in high-ionic-strength alkaline solutions—[NpO2(OH)4]1– or [NpO4(OH)2]3–? Can. J. Chem. 2009, 87, 1436–1443. [Google Scholar] [CrossRef]
- Grigoriev, M.S.; Krot, N.N. Reinvestigation of trisodium dihydroxidotetraoxidoneptunate(VII) dehydrate. Acta Cryst. E 2007, 64, i6. [Google Scholar] [CrossRef] [PubMed]
- Krot, N.N.; Charushnikova, I.A.; Grigor’ev, M.S.; Perminov, V.P. Synthesis and structure of actinide(VII) compounds Rb3NpO4(OH)2·3H2O and Rb3PuO4(OH)2·3H2O. Radiochemistry 2012, 54, 241–246. [Google Scholar] [CrossRef]
- Nikonov, M.V.; Gogolev, A.V.; Tananaev, I.G.; Myasoedov, B.F.; Clark, D.L. Study of Am and Pu behavior in alkaline media. Chimie 2004, 7, 1205–1208. [Google Scholar] [CrossRef]
- Peretroukhine, V.F.; Delegard, C.H. Some Comparisons of Plutonium-Bearing Radwaste Management in the USA and Russia. In The Environmental Challenges of Nuclear Disarmament. NATO Science Series (Series 1: Disarmament Technologies); Baca, T.E., Florkowski, T., Eds.; Springer: Dordrecht, The Netherlands, 2000; Volume 29. [Google Scholar]
- Gelis, A.V.; Vanysek, P.; Jensen, M.P.; Nash, K.L. Electrochemical and spectrophotometric investigations of neptunium in alkaline media. Radiochim. Acta 2001, 89, 565–571. [Google Scholar] [CrossRef]
- Zielen, A.J.; Cohen, D. The neptunium(VII)-(VI) couple in sodium hydroxide solutions. J. Phys. Chem. 1970, 74, 394–405. [Google Scholar] [CrossRef]
- Shilov, V.P.; Krot, N.N.; Gel’man, A.D. Supplementary data on the oxidative properties of neptunium(VII) in alkaline solutions. Radiochim. 1970, 12, 697–699. [Google Scholar]
- Peretrukhin, V.F.; Krot, N.N.; Gel’man, A.D. Formal potentials of the couple Np(VII)-Np(VI) and Pu(VII)-Pu(VI) in aqueous solutions with a high concentration of alkali. Sov. Radiochem. 1972, 14, 68–72. [Google Scholar]
- Ermakov, V.S.; Peretrukhin, V.F.; Krot, N.N. Redox potentials of the couples Np(VI)-Np(V) and Pu(VI)-Pu(V) in LiOH solutions. Sov. Radiochem. 1977, 19, 212–213. [Google Scholar]
- Gaona, X.; Fellhauer, D.; Altmaier, M. Thermodynamic description of Np(VI) solubility, hydrolysis, and redox behavior in dilute to concentrated alkaline NaCl solutions. Pure Appl. Chem. 2013, 85, 2027–2049. [Google Scholar] [CrossRef]
- Fellhauer, D.; Gaona, X.; Rothe, J.; Altmaier, M.; Fanghänel, T. Neptunium(VI) solubility in alkaline CaCl2 solutions: Evidence for the formation of calcium neptunates CaxNpO3+x (s,hyd). Monatsh. Chem. Chem. Mon. 2018, 149, 237–252. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Spitsyn, V.I.; Gelman, A.D.; Krot, N.N.; Mefodiye, M.P.; Zakharov, F.A.; Komkov, Y.A.; Shilov, V.P.; Smirnova, I.V. Heptavalent state of neptunium and plutonium. J. Inorg. Nucl. Chem. 1969, 31, 2733. [Google Scholar] [CrossRef]
- Shilov, V.P.; Stepanov, E.S.; Krot, N.N. Behavior of Neptunium(VII) in carbonate solutions. Sov. Radiochem. 1976, 18, 310–313. [Google Scholar]
- Clark, D.L.; Conradson, S.D.; Donohoe, R.J.; Gordon, P.L.; Webster Keogh, D.; Palmer, P.D.; Scott, B.L.; Tait, C.D. Chemical Speciation of Neptunium(VI) under Strongly Alkaline Conditions. Structure, Composition, and Oxo Ligand Exchange. Inorg. Chem. 2013, 52, 3547–3555. [Google Scholar] [CrossRef]
- Gaona, X.; Wieland, E.; Tits, J.; Scheinost, A.C.; Dähn, R. Np(V/VI) redox chemistry in cementitious systems: XAFS investigations on the speciation under anoxic and oxidizing conditions. Appl. Geochem. 2013, 28, 109–118. [Google Scholar] [CrossRef]
- Status and Trends in Spent Fuel and Radioactive Waste Management; IAEA Nuclear Energy Series No. NW-T-1.14; International Atomic Energy Agency: Vienna, Austria, 2018.
- Neeb, K.-H. The Radiochemistry of Nuclear Power Plants with Light Water Reactors; De Gruyter: Berlin, Germany, 1997. [Google Scholar]
- Kleykamp, H. The chemical state of the fission products in oxide fuels. J. Nucl. Mater. 1985, 131, 221–246. [Google Scholar] [CrossRef]
- Metz, V.; Geckeis, H.; González-Robles, E.; Loida, A.; Bube, C.; Kienzler, B. Radionuclide behaviour in the near-field of a geological repository for spent nuclear fuel. Radiochim. Acta 2012, 100, 699–713. [Google Scholar] [CrossRef]
- Degueldre, C.; Martin, M.; Kuri, G.; Grolimund, D.; Borca, C. Plutonium–uranium mixed oxide characterization by coupling micro-X-ray diffraction and absorption investigations. J. Nucl. Mater. 2011, 416, 142–150. [Google Scholar] [CrossRef]
- Degueldre, C.; Cozzo, C.; Martin, M.; Grolimund, D.; Mieszczynski, C. Americium characterization by X-ray fluorescence and absorption spectroscopy in plutonium uranium mixed oxide. J. Solid State Chem. 2013, 202, 315–319. [Google Scholar] [CrossRef]
- Degueldre, C.; Borca, C.; Cozzo, C. Curium analysis in plutonium uranium mixed oxide by x-ray fluorescence and absorption fine structure spectroscopy. Talanta 2013, 115, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Degueldre, C.; Mieszczynski, C.; Borca, C.; Grolimund, D.; Martin, M.; Bertsch, J. X-ray fluorescence and absorption analysis of krypton in irradiated nuclear fuel. Nucl. Instrum. Methods B 2014, 336, 116–122. [Google Scholar] [CrossRef]
- Degueldre, C.; Pin, S.; Poonoosamy, J.; Kulik, D.A. Redox state of plutonium in irradiated mixed oxide fuels. J. Phys. Chem. Solids 2014, 75, 358–365. [Google Scholar] [CrossRef]
- Degueldre, C.; Bertsch, J.; Martin, M. Post irradiation examination of nuclear fuel: Toward a complete Analysis. Prog. Nucl. Energy 2016, 92, 242–253. [Google Scholar] [CrossRef]
- Curti, E.; Puranen, A.; Grolimund, D.; Jädernas, D.; Sheptyakova, D.; Mesbah, A. Characterization of selenium in UO2 spent nuclear fuel by micro X-ray absorption spectroscopy and its thermodynamic stability. Environ. Sci. Process. Impacts 2015, 17, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Dagan, R.; Herm, H.; Metz, V.; Becker, B. Determination of minor actinides in irradiated fuel rod components. ATW Atomwirtschaft-Atomtechnik Int. J. Nucl. Power 2018, 63, 526–528. [Google Scholar]
- Pelowitz, D. MCNPX User’s Manual; Version 2.7.0, LA-CP-11-00438; LANL: Los Alamos, NM, USA, 2011. [Google Scholar]
- Wilson, W.B.; England, T.R.; George, D.C.; Muir, D.W.; Young, P.G. Recent Development of the CINDER ’90 Transmutation Code and Data Library for Actinide Transmutation Studies. In Proceedings of the GLOBAL ’95 International Conference on Evaluation of Emerging Nuclear Fuel Cycle Systems, Versailles, France, 11–14 September 1995. [Google Scholar]
- Brendebach, B.; Banik, N.L.; Marquardt, C.M.; Rothe, J.; Denecke, M.A.; Geckeis, H. X-ray absorption spectroscopic study of trivalent and tetravalent actinides in solution at varying pH values. Radiochim. Acta 2009, 97, 701–708. [Google Scholar] [CrossRef]
- Bunker, G. Introduction to XAFS; Cambridge University Press: Cambridge, UK, 2010; ISBN 9780511809194. [Google Scholar]
- Li, P.; Chen, I.-W.; Penner-Hahn, J.E. X-ray-absorption studies of zirconia polymorphs. I. Characteristic local structures. Phys. Rev. B 1993, 48, 10063–10073. [Google Scholar] [CrossRef]
- Bès, R.; Rivenet, M.; Solari, P.L.; Kvashnina, K.O.; Scheinost, A.C.; Martin, P.M. Use of HERFD-XANES at the UL3- and M4-Edges To Determine the Uranium Valence State on [Ni(H2O)4]3 [U(OH,H2O)(UO2)8O12(OH)3]. Inorg. Chem. 2016, 55, 4260–4270. [Google Scholar] [CrossRef]
- Podkovyrina, Y.; Pidchenko, I.; Prüßmann, T.; Bahl, S.; Göttlicher, J.; Soldatov, A.; Vitova, T. Probing Covalency in the UO3 Polymorphs by U M4 edge HR- XANES. J. Phys. Conf. Ser. 2016, 712, 012092. [Google Scholar] [CrossRef]
- Walshe, A.; Prussmann, T.; Vitova, T.; Baker, R.J. An EXAFS and HR-XANES study of the uranyl peroxides [UO2(η2-O2)(H2O)2]·nH2O (n = 0, 2) and uranyl(oxy)hydroxide [(UO2)4O(OH)6]·6H2O. Dalton Trans. 2014, 43, 4400–4407. [Google Scholar] [CrossRef]
- Vitova, T.; Green, J.C.; Denning, R.G.; Löble, M.; Kvashnina, K.; Kas, J.J.; Jorissen, K.; Rehr, J.J.; Malcherek, T.; Denecke, M.A. Polarization Dependent High Energy Resolution X-ray Absorption Study of Dicesium Uranyl Tetrachloride. Inorg. Chem. 2015, 54, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Dodge, C.J.; Francis, A.J.; Gillow, J.B.; Halada, G.P.; Eng, C.; Clayton, C.R. Association of uranium with iron oxides typically formed on corroding steel surfaces. Environ. Sci. Technol. 2002, 36, 3504–3511. [Google Scholar] [CrossRef] [PubMed]
- Duff, M.C.; Coughlin, J.U.; Hunter, D.B. Uranium co-precipitation with iron oxide minerals. Geochim. Cosmochim. Acta 2002, 66, 3533–3547. [Google Scholar] [CrossRef]
- O’Loughlin, E.J.; Kelly, S.D.; Cook, R.E.; Csencsits, R.; Kemner, K.M. Reduction of Uranium(VI) by mixed iron(II/iron(III) hydroxide (green rust): Formation of UO2 manoparticies. Environ. Sci. Technol. 2003, 37, 721–727. [Google Scholar] [CrossRef]
- Latta, D.E.; Gorski, C.A.; Boyanov, M.I.; O’Loughlin, E.J.; Kemner, K.M.; Scherer, M.M. Influence of Magnetite Stoichiometry on U-VI Reduction. Environ. Sci. Technol. 2012, 46, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Ilton, E.S.; Boily, J.F.; Buck, E.C.; Skomurski, F.N.; Rosso, K.M.; Cahill, C.L.; Bargar, J.R.; Felmy, A.R. Influence of Dynamical Conditions on the Reduction of U-VI at the Magnetite-Solution Interface. Environ. Sci. Technol. 2010, 44, 170–176. [Google Scholar] [CrossRef]
- Massey, M.S.; Lezama-Pacheco, J.S.; Jones, M.E.; Ilton, E.S.; Cerrato, J.M.; Bargar, J.R.; Fendorf, S. Competing retention pathways of uranium upon reaction with Fe(II). Geochim. Cosmochim. Acta. 2014, 142, 166–185. [Google Scholar] [CrossRef]
- Marshall, T.A.; Morris, K.; Law, G.T.W.; Mosselmans, J.F.W.; Bots, P.; Roberts, H.; Shaw, S. Uranium fate during crystallization of magnetite from ferrihydrite in conditions relevant to the disposal of radioactive waste. Mineral. Mag. 2015, 79, 1265–1274. [Google Scholar] [CrossRef]
- Pidchenko, I.; Kvashnina, K.O.; Yokosawa, T.; Finck, N.; Bahl, S.; Schild, D.; Polly, R.; Bohnert, E.; Rossberg, A.; Göttlicher, J.; et al. Uranium Redox Transformations after U(VI) Coprecipitation with Magnetite Nanoparticles. Environ. Sci. Technol. 2017, 51, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses; Wiley: Hoboken, NJ, USA, 2003. [Google Scholar]
- Seah, M.P.; Gilmore, L.S.; Beamson, G. XPS: Binding energy calibration of electron spectrometers 5—Re-evaluation of the reference energies. Surf. Interface. Anal. 1998, 26, 642–649. [Google Scholar] [CrossRef]
- Glatzel, P.; Bergmann, U. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—Electronic and structural information. Coord. Chem. Rev. 2005, 249, 65–95. [Google Scholar] [CrossRef]
- Belai, N.; Frisch, M.; Ilton, E.S.; Ravel, B.; Cahill, C.L. Pentavalent Uranium Oxide via Reduction of [UO2]2+ under Hydrothermal Reaction Conditions. Inorg. Chem. 2008, 47, 10135–10140. [Google Scholar] [CrossRef] [PubMed]
- Kvashnina, K.O.; Butorin, S.M.; Martin, P.; Glatzel, P. Chemical State of Complex Uranium Oxides. Phys. Rev. Lett. 2013, 111, 253002. [Google Scholar] [CrossRef]
- Huber, F.; Schild, D.; Vitova, T.; Rothe, J.; Kirsch, R.; Schäfer, T. U(VI) removal kinetics in presence of synthetic magnetite nanoparticles. Geochim. Cosmochim. Acta 2012, 96, 154–173. [Google Scholar] [CrossRef]
- Soldatov, A.V.; Lamoen, D.; Konstantinovic, M.J.; Van den Berghe, S.; Scheinost, A.C.; Verwerft, M. Local structure and oxidation state of uranium in some ternary oxides: X-ray absorption analysis. J. Solid State Chem. 2007, 180, 54–61. [Google Scholar] [CrossRef]
- Vitova, T.; Kvashnina, K.O.; Nocton, G.; Sukharina, G.; Denecke, M.A.; Butorin, S.M.; Mazzanti, M.; Caciuffo, R.; Soldatov, A.; Behrends, T.; et al. High energy resolution X-ray absorption spectroscopy study of uranium in varying valence states. Phys. Rev. B 2010, 82, 235118. [Google Scholar] [CrossRef]
- Vitova, T.; Pidchenko, I.; Biswas, S.; Beridze, G.; Dunne, P.W.; Schild, D.; Wang, Z.; Kowalski, P.M.; Baker, R.J. Dehydration of the Uranyl Peroxide Studtite, [UO2(η2-O2)(H2O)2]·2H2O, Affords a Drastic Change in the Electronic Structure: A Combined X-ray Spectroscopic and Theoretical Analysis. Inorg. Chem. 2018, 57, 1735–1743. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Matrix Solution | E’ (mV) |
|---|---|---|
| Zielen and Cohen (1970) [38] | 1.0 M NaOH | 582 |
| Shilov et al. (1970) [39] | 1.0 M NaOH | 587 |
| Peretrukhin et al. (1972) [40] | 3.0 M NaOH 11.0 M NaOH 14.1 M NaOH | 497 209 131 |
| Ermakov et al. (1977) [41] | 1.0 M LiOH | 604 |
| Oxid. State | Reference | Matrix Solution | N | R [Å] | σ2 [Å2] | ΔE0 [eV] |
|---|---|---|---|---|---|---|
| Np(VII) | This work a | 2.0 M NaOH + 3.0 M NaCl | O1: 4 * O2: 3.4 ± 0.9 | 1.90 ± 0.005 2.32 ± 0.01 | 0.0008 ± 0.0005 0.0073 ± 0.003 | 6.1 ± 1.3 |
| Williams et al. (2001) [27] b | 1.0 M NaOH | O1: 4 ± 1 O2: 2.4 ± 0.6 Na: 2 ± 1 | 1.87 ± 0.01 2.24 ± 0.04 2.34 ± 0.04 | 0.003 ± 0.001 | 6.2 ± 0.9 | |
| Bolvin et al. (2001) [28] c | 2.5 M NaOH | O1: 3.6 ± 0.3 O2: 3.3 ± 1.3 | 1.89 ± 0.04 2.32 ± 0.06 | 0.0020 0.0133 | –5.0 | |
| Ikeda-Ohno et al. (2009) [30] d | 2.5 M NaOH | O1: 4.0 ± 0.4 O2: 2.4 ± 0.2 OMS: 4.0 ± 0.4 | 1.89 ± 0.01 2.35 ± 0.01 3.79 ± 0.01 | 0.0023 ± 0.0005 0.0073 ± 0.0005 0.0074 ± 0.0005 | 6.81 | |
| Np(VI) | Clark et al. (2013) [47] e | 2.5 M TMA-OH | O1: 2.0 O2: 3.8 | 1.80 ± 0.01 2.23 ± 0.01 | 0.0055 * 0.0088 | 2.50 0.76 |
| Gaona et al. (2013) [48] f | 0.5 M TMA-OH | O1: 1.8 ± 0.4 O2: 4.6 ± 0.8 | 1.80 ± 0.01 2.25 ± 0.02 | 0.001 ± 0.001 0.005 ± 0.002 | 10.4 ± 2.0 |
| Element | Th | U | Np | Pu | Am | Cm | Zr |
|---|---|---|---|---|---|---|---|
| total at.% | 6.27 × 10−7 | 30.9 | 2.61 × 10−2 | 0.370 | 5.45 × 10−2 | 1.62 × 10−3 | 0.412 |
| Short Name | Contact Time, Days | U Removed, % | pH | Eh, mV | Technique |
|---|---|---|---|---|---|
| Um175 | 175 | 99.5 | 7.5 | 50 | U M4/L3 edge HR-XANES, U 4f, Fe 2p XPS |
| Um175f | 175 + 2 days after adding Fe(II) | 99.5 | 7.1 | −150 | U M4/L3 edge HR-XANES, U 4f, Fe 2p XPS |
| Umh | 55 | 99 | 7.7 | 160 | U M4/L3 edge HR-XANES |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rothe, J.; Altmaier, M.; Dagan, R.; Dardenne, K.; Fellhauer, D.; Gaona, X.; González-Robles Corrales, E.; Herm, M.; Kvashnina, K.O.; Metz, V.; et al. Fifteen Years of Radionuclide Research at the KIT Synchrotron Source in the Context of the Nuclear Waste Disposal Safety Case. Geosciences 2019, 9, 91. https://doi.org/10.3390/geosciences9020091
Rothe J, Altmaier M, Dagan R, Dardenne K, Fellhauer D, Gaona X, González-Robles Corrales E, Herm M, Kvashnina KO, Metz V, et al. Fifteen Years of Radionuclide Research at the KIT Synchrotron Source in the Context of the Nuclear Waste Disposal Safety Case. Geosciences. 2019; 9(2):91. https://doi.org/10.3390/geosciences9020091
Chicago/Turabian StyleRothe, Jörg, Marcus Altmaier, Ron Dagan, Kathy Dardenne, David Fellhauer, Xavier Gaona, Ernesto González-Robles Corrales, Michel Herm, Kristina O. Kvashnina, Volker Metz, and et al. 2019. "Fifteen Years of Radionuclide Research at the KIT Synchrotron Source in the Context of the Nuclear Waste Disposal Safety Case" Geosciences 9, no. 2: 91. https://doi.org/10.3390/geosciences9020091
APA StyleRothe, J., Altmaier, M., Dagan, R., Dardenne, K., Fellhauer, D., Gaona, X., González-Robles Corrales, E., Herm, M., Kvashnina, K. O., Metz, V., Pidchenko, I., Schild, D., Vitova, T., & Geckeis, H. (2019). Fifteen Years of Radionuclide Research at the KIT Synchrotron Source in the Context of the Nuclear Waste Disposal Safety Case. Geosciences, 9(2), 91. https://doi.org/10.3390/geosciences9020091