Foreign or Domestic CARs: Receptor Ligands as Antigen-Binding Domains

{kind=link}
Abstract
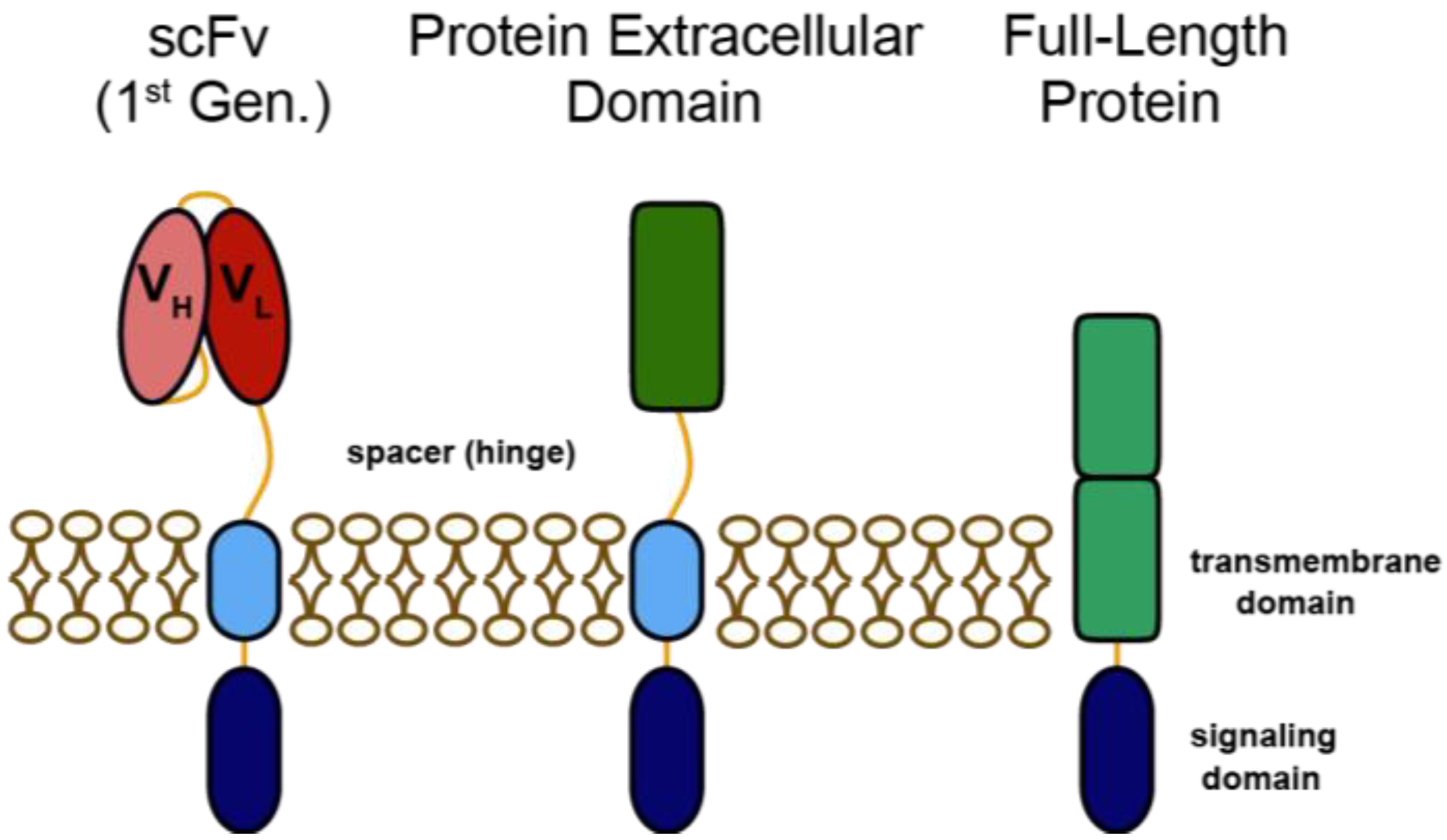
:1. Introduction

2. Heregulin-ζ CARs for Redirecting T Cells to HER3 and HER4
3. Targeting the Tumor Vasculature: Vascular Endothelial Growth Factor CARs
4. IL13 Zetakine T Cells for the Treatment of Glioblastoma
5. Targeting NKG2D Ligands with NKG2D CARs
6. Immunotherapy of CD70-Positive Malignancies with a CD27-ζ CAR
7. Peptides as CAR-Binding Domains
8. Conclusions
Acknowledgements
Author Contributions
Conflicts of interest
References
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Canc. Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar]
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J. Gene Med. 2012, 14, 405–415. [Google Scholar]
- Maus, M.V.; Haas, A.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Carraway, K.L., 3rd; Cantley, L.C. A neu acquaintance for erbB3 and erbB4: A role for receptor heterodimerization in growth signaling. Cell 1994, 78, 5–8. [Google Scholar] [CrossRef]
- Altenschmidt, U.; Kahl, R.; Moritz, D.; Schnierle, B.S.; Gerstmayer, B.; Wels, W.; Groner, B. Cytolysis of tumor cells expressing the Neu/erbB-2, erbB-3, and erbB-4 receptors by genetically targeted naive T lymphocytes. Clin. Canc. Res. 1996, 2, 1001–1008. [Google Scholar]
- Muniappan, A.; Banapour, B.; Lebkowski, J.; Talib, S. Ligand-mediated cytolysis of tumor cells: Use of heregulin-zeta chimeras to redirect cytotoxic T lymphocytes. Canc. Gene Ther. 2000, 7, 128–134. [Google Scholar]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. New Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist 2004, 9, 2–10. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Fehrenbacher, L.; Hainsworth, J.D.; Heim, W.; Berlin, J.; Holmgren, E.; Hambleton, J.; Novotny, W.F.; Kabbinavar, F. Bevacizumab in combination with fluorouracil and leucovorin: An active regimen for first-line metastatic colorectal cancer. J. Clin. Oncol. 2005, 23, 3502–3508. [Google Scholar] [CrossRef]
- Shojaei, F. Anti-angiogenesis therapy in cancer: Current challenges and future perspectives. Canc. Lett. 2012, 320, 130–137. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Li, L.; Sanoff, H.K.; Carpenter, W.T.; Schrag, D. Effectiveness of bevacizumab with first-line combination chemotherapy for Medicare patients with stage IV colorectal cancer. J. Clin. Oncol. 2012, 30, 608–615. [Google Scholar] [CrossRef]
- Niederman, T.M.; Ghogawala, Z.; Carter, B.S.; Tompkins, H.S.; Russell, M.M.; Mulligan, R.C. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 7009–7014. [Google Scholar] [CrossRef]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Salsman, V.S.; Chow, K.K.; Shaffer, D.R.; Kadikoy, H.; Li, X.N.; Gerken, C.; Perlaky, L.; Metelitsa, L.S.; Gao, X.; Bhattacharjee, M.; et al. Crosstalk between medulloblastoma cells and endothelium triggers a strong chemotactic signal recruiting T lymphocytes to the tumor microenvironment. PLoS One 2011, 6, e20267. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M.; Hulet, S.W.; Connor, J.R.; Gillespie, G.Y. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin. Canc. Res. 1999, 5, 985–990. [Google Scholar]
- Hilton, D.J.; Zhang, J.G.; Metcalf, D.; Alexander, W.S.; Nicola, N.A.; Willson, T.A. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 497–501. [Google Scholar]
- Debinski, W.; Gibo, D.M.; Obiri, N.I.; Kealiher, A.; Puri, R.K. Novel anti-brain tumor cytotoxins specific for cancer cells. Nat. Biotechnol. 1998, 16, 449–453. [Google Scholar] [CrossRef]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Canc. Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef]
- Brown, C.E.; Starr, R.; Naranjo, A.; Wright, C.; Bading, J.; Ressler, J.A.; D’Apuzzo, M.; Badie, B.; Forman, S.J.; Jensen, C.J. Adoptive Transfer of Autologous IL13-zetakine positive Engineered T Cell Clones for the Treatment of Recurrent Glioblastoma: Lessons from the clinic. Mol. Ther. 2011, 19, S136–S137. [Google Scholar]
- Kong, S.; Sengupta, S.; Tyler, B.; Bais, A.J.; Ma, Q.; Doucette, S.; Zhou, J.; Sahin, A.; Carter, B.S.; Brem, H.; et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin. Canc. Res. 2012, 18, 5949–5960. [Google Scholar] [CrossRef]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef]
- Jinushi, M.; Hodi, F.S.; Dranoff, G. Therapy-induced antibodies to MHC class I chain-related protein A antagonize immune suppression and stimulate antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2006, 103, 9190–9195. [Google Scholar] [CrossRef]
- Zhang, T.; Lemoi, B.A.; Sentman, C.L. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood 2005, 106, 1544–1551. [Google Scholar] [CrossRef]
- Barber, A.; Zhang, T.; DeMars, L.R.; Conejo-Garcia, J.; Roby, K.F.; Sentman, C.L. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Canc. Res. 2007, 67, 5003–5008. [Google Scholar] [CrossRef]
- Zhang, T.; Barber, A.; Sentman, C.L. Chimeric NKG2D modified T cells inhibit systemic T-cell lymphoma growth in a manner involving multiple cytokines and cytotoxic pathways. Canc. Res. 2007, 67, 11029–11036. [Google Scholar] [CrossRef]
- Barber, A.; Meehan, K.R.; Sentman, C.L. Treatment of multiple myeloma with adoptively transferred chimeric NKG2D receptor-expressing T cells. Gene Ther. 2011, 18, 509–516. [Google Scholar] [CrossRef]
- Barber, A.; Zhang, T.; Megli, C.J.; Wu, J.; Meehan, K.R.; Sentman, C.L. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. Exp. Hematol. 2008, 36, 1318–1328. [Google Scholar] [CrossRef]
- Zhang, T.; Barber, A.; Sentman, C.L. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Canc. Res. 2006, 66, 5927–5933. [Google Scholar] [CrossRef]
- Song, D.G.; Ye, Q.; Santoro, S.; Fang, C.; Best, A.; Powell, D.J., Jr. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum. Gene Ther. 2013, 24, 295–305. [Google Scholar] [CrossRef]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011, 121, 1822–1826. [Google Scholar] [CrossRef]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef]
- Denoeud, J.; Moser, M. Role of CD27/CD70 pathway of activation in immunity and tolerance. J. Leukoc. Biol. 2011, 89, 195–203. [Google Scholar] [CrossRef]
- Arens, R.; Nolte, M.A.; Tesselaar, K.; Heemskerk, B.; Reedquist, K.A.; van Lier, R.A.; van Oers, M.H. Signaling through CD70 regulates B cell activation and IgG production. J. Immunol. 2004, 173, 3901–3908. [Google Scholar]
- Agathanggelou, A.; Niedobitek, G.; Chen, R.; Nicholls, J.; Yin, W.; Young, L.S. Expression of immune regulatory molecules in Epstein-Barr virus-associated nasopharyngeal carcinomas with prominent lymphoid stroma. Evidence for a functional interaction between epithelial tumor cells and infiltrating lymphoid cells. Am. J. Pathol. 1995, 147, 1152–1160. [Google Scholar]
- Hunter, Z.R.; Branagan, A.R.; Santos, D.D.; Tournilhac, O.; Hatjiharissi, E.; Xu, L.; Manning, R.J.; Treon, S.P. High levels of soluble immunoregulatory receptors in patients with Waldenstrom’s macroglobulinemia. ASH Annual Meeting Abstracts. 2004, 104, 4881. [Google Scholar]
- Lens, S.M.; Drillenburg, P.; den Drijver, B.F.; van Schijndel, G.; Pals, S.T.; van Lier, R.A.; van Oers, M.H. Aberrant expression and reverse signalling of CD70 on malignant B cells. Br. J. Haematol. 1999, 106, 491–503. [Google Scholar] [CrossRef]
- Baba, M.; Okamoto, M.; Hamasaki, T.; Horai, S.; Wang, X.; Ito, Y.; Suda, Y.; Arima, N. Highly enhanced expression of CD70 on human T-lymphotropic virus type 1-carrying T-cell lines and adult T-cell leukemia cells. J. Virol. 2008, 82, 3843–3852. [Google Scholar] [CrossRef]
- McEarchern, J.A.; Smith, L.M.; McDonagh, C.F.; Klussman, K.; Gordon, K.A.; Morris-Tilden, C.A.; Duniho, S.; Ryan, M.; Boursalian, T.E.; Carter, P.J.; Grewal, I.S.; Law, C.L. Preclinical characterization of SGN-70, a humanized antibody directed against CD70. Clin. Cancer Res. 2008, 14, 7763–7772. [Google Scholar] [CrossRef]
- Junker, K.; Hindermann, W.; von, E.F.; Diegmann, J.; Haessler, K.; Schubert, J. CD70: A new tumor specific biomarker for renal cell carcinoma. J. Urol. 2005, 173, 2150–2153. [Google Scholar] [CrossRef]
- Chahlavi, A.; Rayman, P.; Richmond, A.L.; Biswas, K.; Zhang, R.; Vogelbaum, M.; Tannenbaum, C.; Barnett, G.; Finke, J.H. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Canc. Res. 2005, 65, 5428–5438. [Google Scholar] [CrossRef]
- Shaffer, D.R.; Savoldo, B.; Yi, Z.; Chow, K.K.; Kakarla, S.; Spencer, D.M.; Dotti, G.; Wu, M.F.; Liu, H.; Kenney, S.; Gottschalk, S. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood 2011, 117, 4304–4314. [Google Scholar] [CrossRef]
- Shaffer, D.R.; Sheehan, A.M.; Yi, Z.; Rodgers, C.C.; Bollard, C.M.; Brenner, M.K.; Rooney, C.M.; Heslop, H.E.; Gottschalk, S. Aggressive peripheral CD70-positive T-cell lymphoma associated with severe chronic active EBV infection. Pediatr. Blood Canc. 2012, 59, 758–761. [Google Scholar] [CrossRef] [Green Version]
- Song, D.G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J., Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Duong, C.P.; Westwood, J.A.; Yong, C.S.; Murphy, A.; Devaud, C.; John, L.B.; Darcy, P.K.; Kershaw, M.H. Engineering T cell function using chimeric antigen receptors identified using a DNA library approach. PLoS One 2013, 8, e63037. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Dudley, M.E. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr. Opin. Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef]
- Pameijer, C.R.; Navanjo, A.; Meechoovet, B.; Wagner, J.R.; Aguilar, B.; Wright, C.L.; Chang, W.C.; Brown, C.E.; Jensen, M.C. Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T cell antigen receptor. Canc. Gene Ther. 2007, 14, 91–97. [Google Scholar] [CrossRef]
- Van Aarsen, L.A.; Leone, D.R.; Ho, S.; Dolinski, B.M.; McCoon, P.E.; LePage, D.J.; Kelly, R.; Heaney, G.; Rayhorn, P.; Reid, C.; et al. Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Canc. Res. 2008, 68, 561–570. [Google Scholar] [CrossRef]
- Davies, D.M.; Foster, J.; van der Stegen, S.J.; Parente-Pereira, A.C.; Chiapero-Stanke, L.; Delinassios, G.J.; Burbridge, S.E.; Kao, V.; Liu, Z.; Bosshard-Carter, L.; et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012, 18, 565–576. [Google Scholar]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Canc. Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Canc. Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shaffer, D.R.; Zhou, P.; Gottschalk, S. Foreign or Domestic CARs: Receptor Ligands as Antigen-Binding Domains. Med. Sci. 2014, 2, 23-36. https://doi.org/10.3390/medsci2010023
Shaffer DR, Zhou P, Gottschalk S. Foreign or Domestic CARs: Receptor Ligands as Antigen-Binding Domains. Medical Sciences. 2014; 2(1):23-36. https://doi.org/10.3390/medsci2010023
Chicago/Turabian StyleShaffer, Donald R., Penghui Zhou, and Stephen Gottschalk. 2014. "Foreign or Domestic CARs: Receptor Ligands as Antigen-Binding Domains" Medical Sciences 2, no. 1: 23-36. https://doi.org/10.3390/medsci2010023
APA StyleShaffer, D. R., Zhou, P., & Gottschalk, S. (2014). Foreign or Domestic CARs: Receptor Ligands as Antigen-Binding Domains. Medical Sciences, 2(1), 23-36. https://doi.org/10.3390/medsci2010023