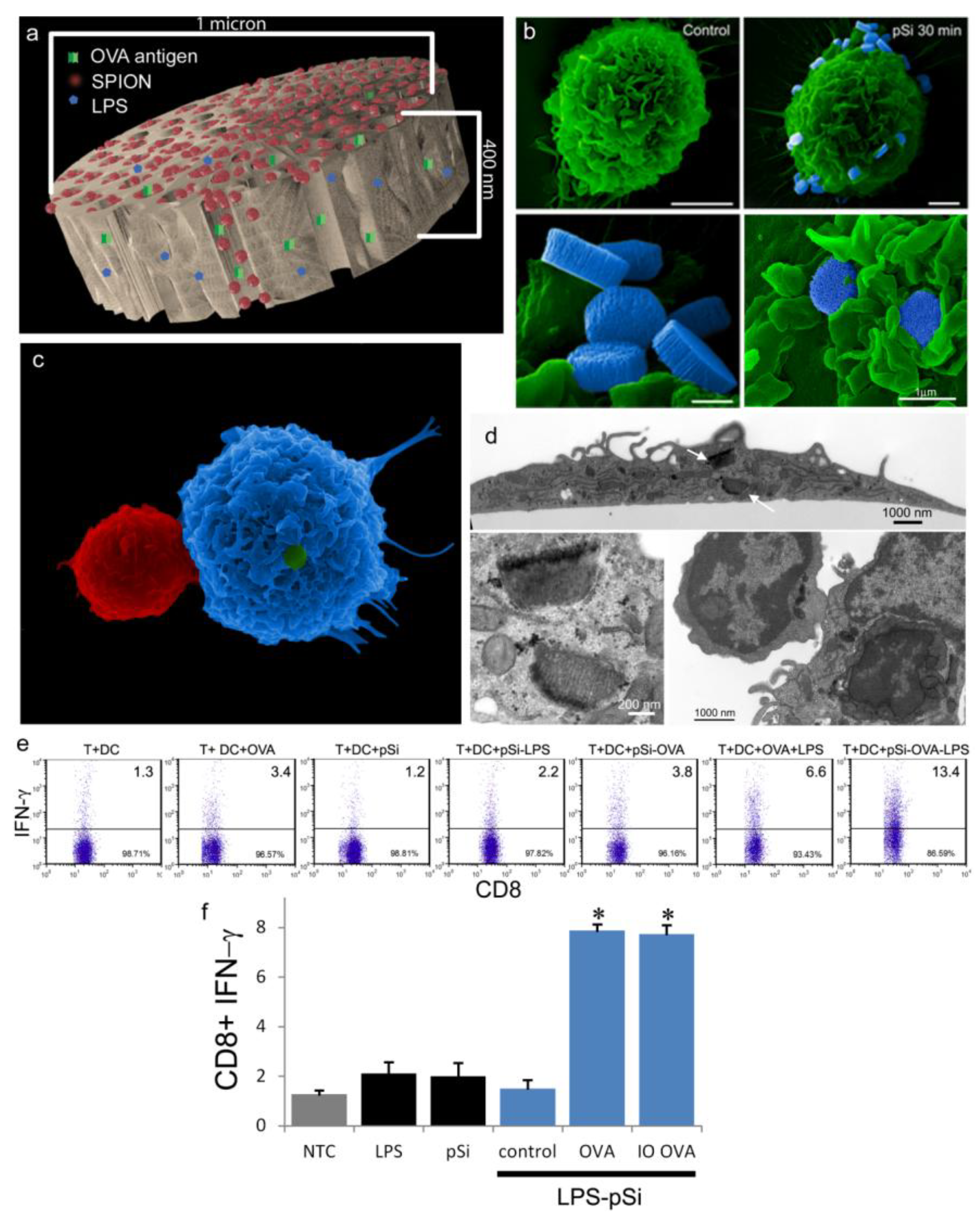
1. Introduction
Cellular uptake of microbes by dendritic cells (DC) is accompanied by engagement of pattern recognition receptors [
1,
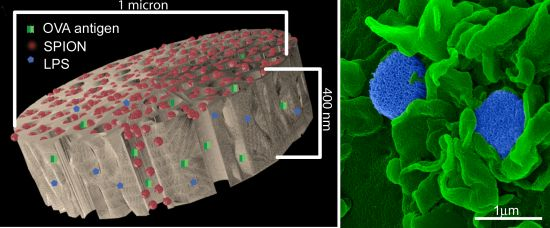
2]. Activation of these receptors induces phagocytosis and expression of genes that cause maturation of the cell and activation of anti-microbial events, thereby inducing innate immunity. Particles carrying pathogen-associated molecular patterns, e.g., lipopolysaccharide (LPS), similarly induce phagocytosis and activate antigen presenting cells (APC). We previously demonstrated that DC incubated with porous silicon (pSi) particles presenting LPS and antigen (
i.e., ovalbumin peptide SIINFEKL) are actively engaged by T cells obtained from C57BL/6-Tg(TcraTcrb) (a.k.a. OT-1) mice expressing a transgenic TCR for recognition of major histocompatibility complex (MHC) class I-presented antigen. The stimulated DC have enhanced migration to the draining lymph node and elevated expression of MHC and costimulatory molecules, as well as increased secretion of proinflammatory cytokines. While these early studies relied on
ex vivo modification of APC with fluorescent tracers, particle-based vaccines that incorporate contrast agents can be used experimentally or clinically for noninvasive detection by magnetic resonance imaging (MRI) to track migration of particles, or their cell-based carriers, to lymphatic tissue for modulation of immune responses.
Contrast in MR images is due to differences in signal relaxation rates that are unique to the physical and chemical characteristics of biological tissues [
3]. Return to the equilibrium occupancy of spins following excitation is referred to as longitudinal relaxation, and is characterized by an exponential time constant generally referred to as T
1. Attenuation of the excited signal due to the dephasing of spins is known as transverse relaxation, which is referred to as T
2. Inhomogeneities in the magnetic field that are not compensated for through the use of a spin-echo pulse sequences lead to shorter transverse relaxation time constants, denoted as T
2* to differentiate intrinsic tissue relaxation from that induced by magnetic field inhomogeneities. Exogenous contrast agents act to reduce these time constants; for example, an agent that reduces T
1 increases signal intensity by accelerating the return to equilibrium between excitations, while an agent that reduces T
2 or T
2* causes the signal to attenuate more quickly after excitation, leading to negative (dark) contrast [
4,
5]. The relaxation time (T
2, T
2*) and concentration of contrast agent are related by the following equations [
6]:
where
T1,2 is the relaxation time at a given concentration of contrast agent,
![Medsci 02 00051 i002]()
is the relaxation time of the medium with contrast agent removed,
C is the concentration of contrast agent, and
r1,2 is the concentration-independent relaxivity of the contrast agent under study. The term
R1,2 in the above equation is referred to as the relaxation rate.
Superparamagnetic iron oxide nanoparticles (SPIONs) shorten T
2 and T
2* due to their large magnetic moments and the large magnetic field gradients that surround them [
7]. The ability of SPIONs to enhance contrast depends on their size, surface properties, aggregation and compartmentalization [
8,
9,
10]. For homogeneously dispersed small particles, the motional averaging regime (MAR theory) predicts that transverse relaxivities, r
2 and r
2*, increase with increasing core size [
11]. That is, the ability of SPIONs to act as contrast agents to lower the T
2 relaxation time of tissues is enhanced by increasing the particle core size due to increased magnetic susceptibility. It has been shown that increasing the core size results in a higher induced magnetization at saturation and thus a higher magnetic susceptibility, calculated as
M = χ
• H, where M is the induced magnetization,
H is the field strength of the magnet, and χ is the suseptibility of the magnetic particle [
11,
12]. Above a certain size, further increases in core size fail to enhance relaxation rates based on the existence of strong dipolar fields surrounding the particle, limiting the impact of water diffusion and leading to a plateau in relaxation rate. For smaller particles, r
2 and r
2* should be nearly equal based on dependence on diffusion effects, however, for larger particles, the static dephasing regime (SDR theory) predicts that R
2* increases with increasing local magnetic dose, meaning that particle aggregation leads to increases in R
2* [
8]. In this study, we compare transverse relaxivities of SPIONs with increasing particle core size ranging from 5 to 30 nm; and we examine the impact of SPION confinement within a pSi matrix on magnetic properties [
13]. This data was presented in an abstract in the Proceedings of the ASME 2nd Global Congress on NanoEngineering for Medicine and Biology [
13].
Size and aspect ratio of particles are important properties that impact particle biodistribution, cellular interactions, and cellular internalization [
14,
15,
16]. With respect to subcutaneous inoculation, particles traffic to lymphatic tissue (
i.e., lymph nodes) in a size-dependent manner, with large particles being engulfed by peripheral APCs at the site of injection, and small nanoparticles being internalized by resident APCs following cell-free trafficking [
17]. In this study, we explore accumulation of free and pSi-encapsulated SPIONs in the spleen following intravenous injection to study the impact of nanoparticle and microparticle trafficking to lymphatic tissue via the vascular system. We further compare T cell immunity stimulated by pSi microparticles presented as single verses hybrid particle complexes.
2. Experimental Section
2.1. Materials
SPIONs were purchased from Ocean Nanotech, LLC (Springdale, AR, USA). The iron oxide nanocrystals had an amphiphilic polymer coating containing terminal amine groups attached to polyethylene glycol linkers, or carboxylic acid moieties. C57BL/6 and C57BL/6-Tg(TcraTcrb) (OT‑1) mice were obtained from Charles River Laboratories, Inc. (Wilmington, MA, USA) and The Jackson Laboratory (Bar Harbor, ME, USA), respectively.
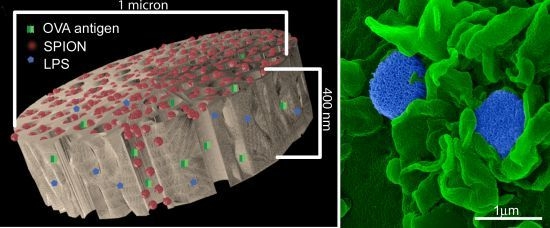
2.2. Fabrication of pSi Microparticles
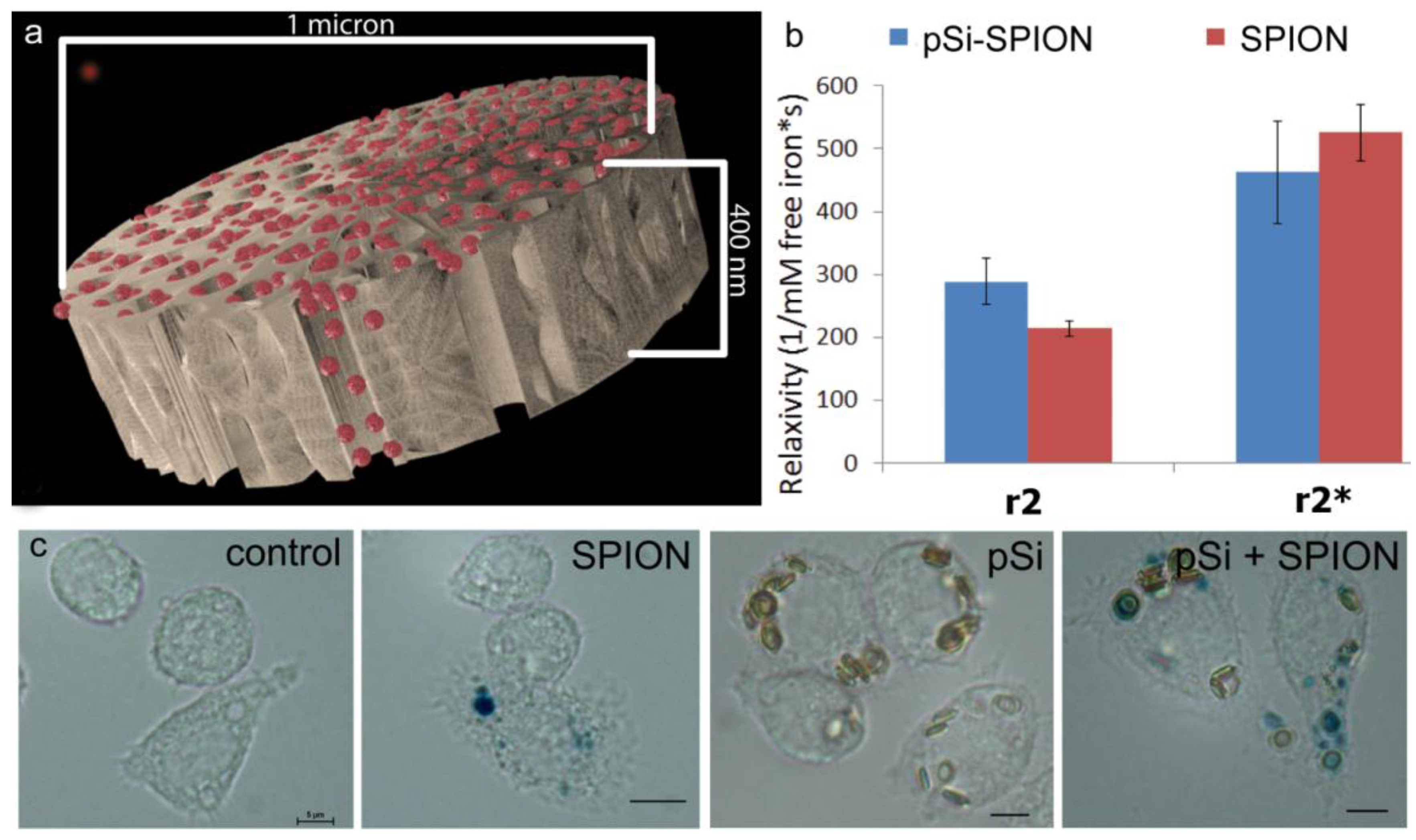
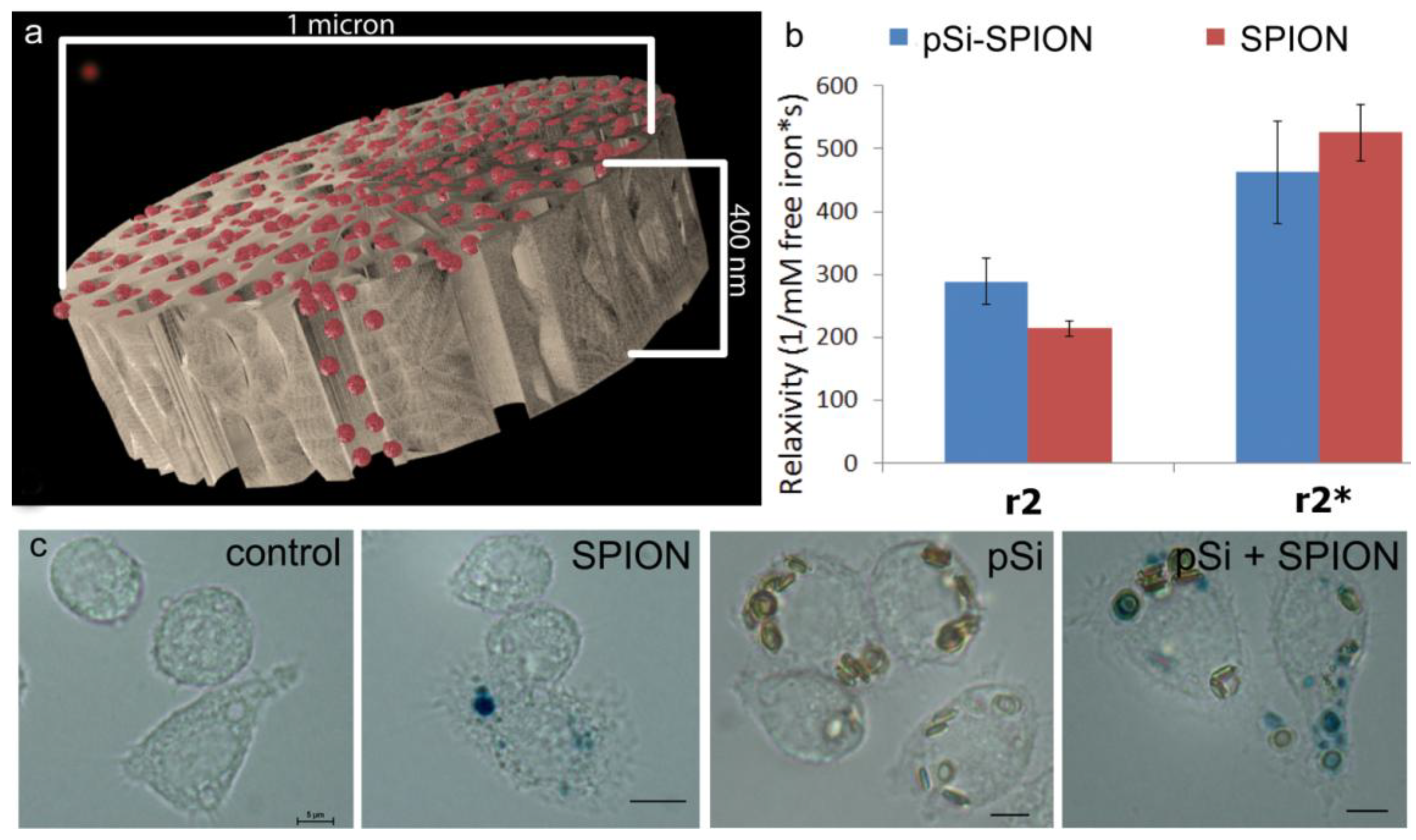
1 µm (diameter) × 400 nm (thickness) discoidal pSi microparticles were fabricated in the Microelectronics Research Center at The University of Texas at Austin by combination of electrochemical etching and standard photolithography. Using heavily doped p++ type (100) silicon wafers with resistivity of 0.005 ohm-cm (Silicon Quest, Inc., Santa Clara, CA, USA) as the silicon source and 1 HF (49%): 3 ethanol solution as etchant, the anodic etching process started with applying a 2.3 mA/cm2 current for 20 s to form a low porosity mechanical layer, then the current was ramped to 14 mA/cm2 in a time course of 15 s, and kept at 14 mA/cm2 for 45 s to form the porous layer with 60 nm average pores. A current of 76 mA/cm2 was applied for 6 s to form the high porosity release layer. Following the formation of porous film, a 40 nm SiO2 layer was deposited by Low Pressure Chemical Vapor Deposition at 400 °C. Standard photolithography was then used to pattern an array of 1 µm circles over the SiO2 capped porous film using a contact aligner (K.Suss MA6 mask aligner) and NR9-500P photoresist (Futurrex, Franklin, NJ, USA). The pattern was transferred into the porous film by Reactive Ion Etch in CF4 plasma (Plasmatherm BatchTop, 15 sccm CF4, 100 mTorr, 200 W RF). The capping SiO2 layer was removed in 49% HF, and the microparticles were released from the substrate in isopropanol by sonication.
2.3. Surface Modification of pSi Microparticles
pSi microparticles were oxidized with piranha solution (1 volume H
2O
2 and 2 volumes of H
2SO
4) with heating to 110–120 °C for 2 h as previously described [
18]. Oxidized pSi microparticles were washed in IPA, and then suspended in IPA containing 0.5% (
v/
v) APTES (Sigma-Aldrich, St. Louis, MO, USA) for 0.5–2 h at 35 °C and 1300 rpm. The APTES-modified pSi microparticles were washed in IPA and dried in a desiccator. Volumetric particle size, size distribution and count were obtained using a Multisizer 4 Coulter
® Particle Counter (Beckman Coulter, Fullerton, CA, USA). The morphology of the particles was verified by scanning electron microscopy (SEM, FEI, Hillsboro, OR, USA).
2.4. MRI Phantoms
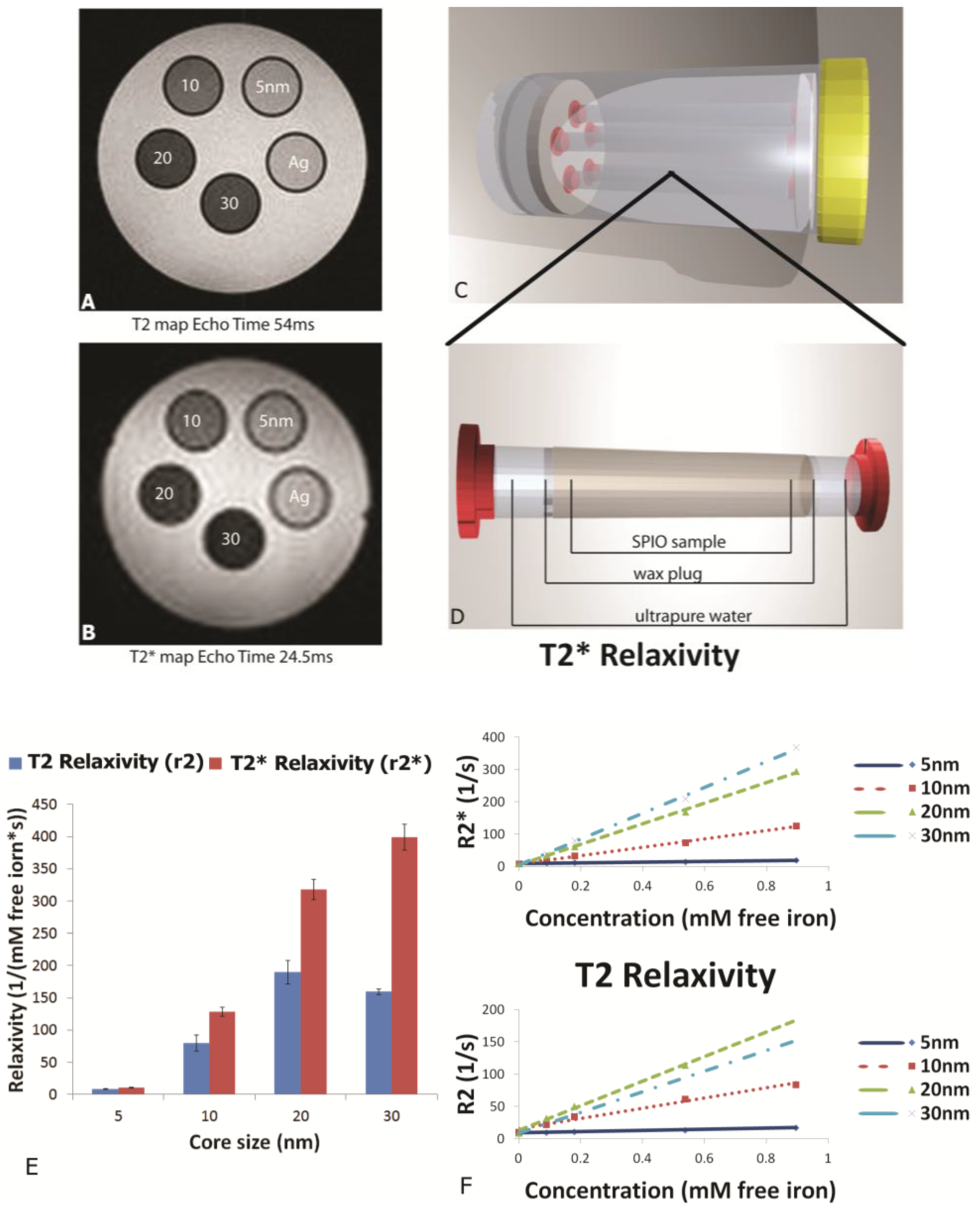
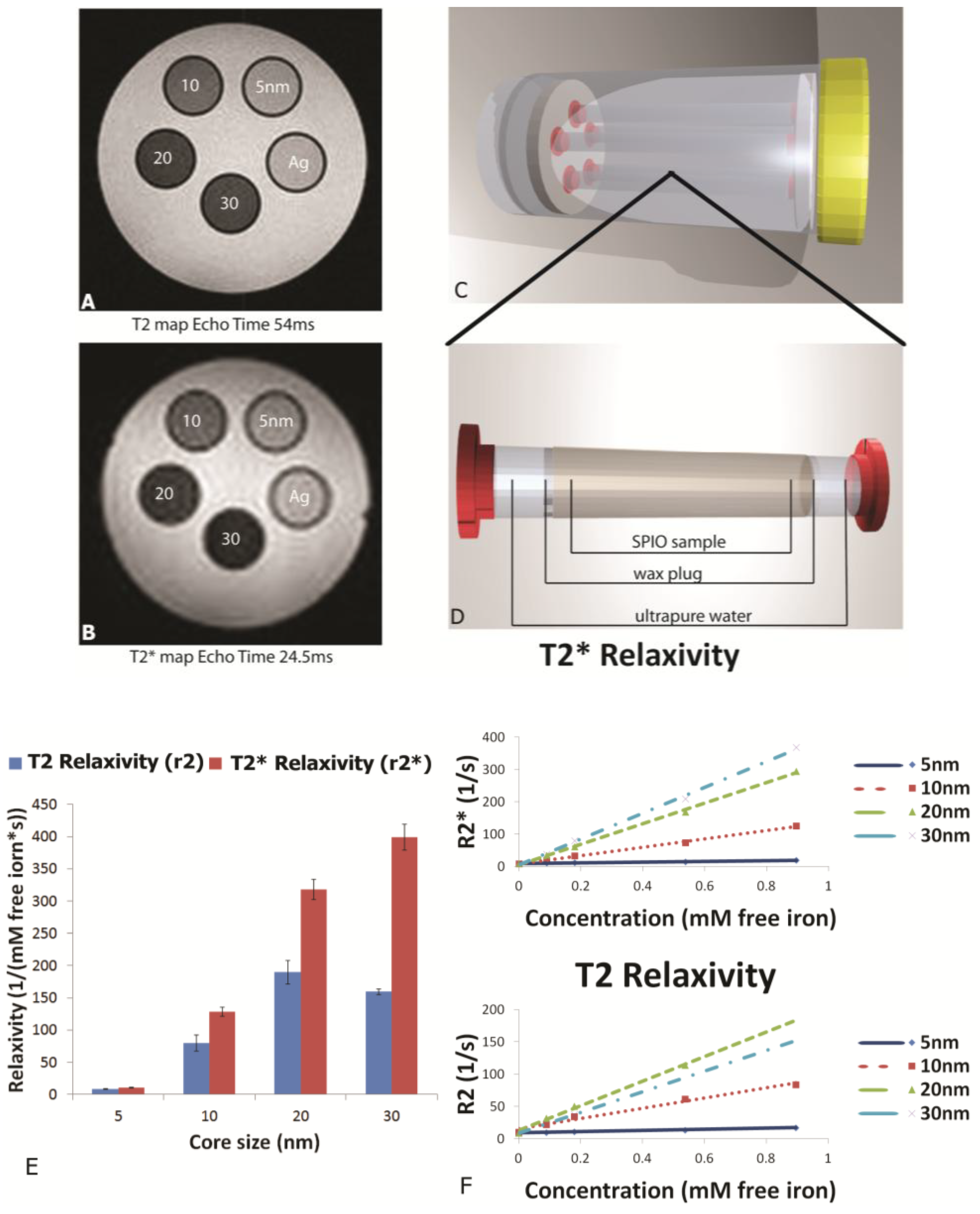
MR phantoms were prepared in thin wall NMR tubes with 10 mm outer diameters (Wilmad Labglass, Vineland, NJ, USA). Samples of free SPIONs with core sizes of 5, 10, 20, and 30 nm were prepared by adding a 0.005, 0.01, 0.03, and 0.05 mg Fe/mL to a 1% agarose suspension to create unique sample tubes for each core size. Iron content was verified using a Prussian blue assay as described later. Sample sets of 4–5 phantoms, including a 1% agarose only control, were moved into a specially-machined tube holder which was placed into a 300 mL cylindrical polypropylene tube. The tube was then filled with relaxed water (doped with 0.1% Magnevist v/v) to reduce proximity to and interference from air interfaces, capped, sealed, and imaged. MR imaging of phantoms was carried out in an actively-shielded 7 Tesla Biospec USR70/30 (Bruker Biospin MRI, Billerica, MA, USA) small animal MRI system equipped with a 30-cm bore, imaging gradients with a 6-cm free bore, and a linear 1H birdcage-style volume resonator with 35 mm inner diameter. A three-plane, T2-weighted fast spin‑echo sequence (TE = 50 ms, TR = 2000 ms, ETL = 8) was used to confirm sample placement. Transverse relaxation time constants (T2*) were measured using a multi-echo gradient-echo sequence (minimum TE = 1.5 ms, echo spacing 3.25 ms, 24 echoes, TR = 4,000 ms, 30 excitation).
2.5. Loading SPIONs into pSi Microparticles
Experiments comparing relaxivity values for free verses pSi-encapsulated 30 nm SPIONs were performed by loading 1 × 108 pSi microparticles with serial dilutions of 5, 2.5, 1.75 mg Fe/mL. Loading was accomplished by means of capillary action by adding SPION suspensions to dry pSi microparticles, briefly sonicating, and incubating for 15 min at room temperature. The microparticles were then washed with 50 µL of DI water and centrifuged at 5000× g × 10 min. The supernatant with excess unloaded SPIONs was pipetted off and the pSi microparticles were desiccated under vacuum. The loading and wash steps were then repeated and the resulting loaded pSi microparticles were suspended in 1% agarose for imaging. The iron content in the samples was verified using inductively coupled plasma-atomic emission spectroscopy (ICP-AES, Agilent Technologies, Santa Clara, CA, USA) as described below.
2.6. ICP-AES Analysis of Iron in Phantoms and Tissue Samples
To verify the iron content of the resected tissues, spleen were dehydrated with ethanol and dried overnight. Samples were weighed and placed into nickel crucibles, which were heated in a 500 °C oven for several hours. Once fully ashed, tissue remains were collected by washing the crucibles with 1% spectrosol™ solution and sonicating, up to a final volume of 5 mL. Samples were centrifuged at 4200 rpm for 10 min to remove ash aggregates from the solution and 4 mL of the dissolved sample was moved to a new conical tube. 1mL of a 1% spectrosol solution, plus 50 μL of 100 mg/L yttrium standard solution, was added to each tube and were analyzed using iron standard concentrations of 25, 75, 100, 250, and 500 ppb, one blank sample of 1% spectrosol solution, and one quality control internal standard containing 250 ppb of iron. Samples were analyzed with a Varian Vista AX at a power of 1 kW, plasma flow set to 15 L/min, auxiliary flow of 1.5 L/min, and a nebulizer flow of 0.75 L/min, with 5 replicate readings at 15 s between each reading. The concentration of iron in agarose phantoms was measured by serial dilution of the phantom contents with a reference range of 25–500 ppb. A final volume of 5 mL of 1% spectrosol plus sample solution was used.
2.7. Prussian Blue Assay for Iron Content
To verify total iron content of phantoms used for particle size comparisons, 5 µL samples of 5, 10, 20, and 30 nm stock solutions, as well as Ferric iron (III) standards at 1.5, 1, 0.5, 0.1, and 0.05 mg/mL were incubated with 120 µL 6 N HCl for 2 h at 60 °C in triplicate. Iron was then oxidized using 0.1 mg/mL ammonium persulfate (BioRad, Richmond, CA, USA) and the color reaction was initiated by adding 125 μL of a 5% K4[Fe(CN)6]·3H2O (Sigma-Aldrich) for 10 min. Following incubation, sample absorbance was determined at 690 nm using a SPECTRA max M2 plate reader (Molecular Devices, Sunnyvale, CA, USA). A standard curve was then generated using iron III hexahydrate (Sigma-Aldrich) and the iron concentration of SPION preparations was calculated. Cells treated with particles for 24 h (20 pSi particles/cell or 2 µg/mL Fe) were labeled by incubation of cells in acid solution containing potassium ferrocyanide for 30 min at 37 °C.
2.8. In Vivo Imaging Experiments
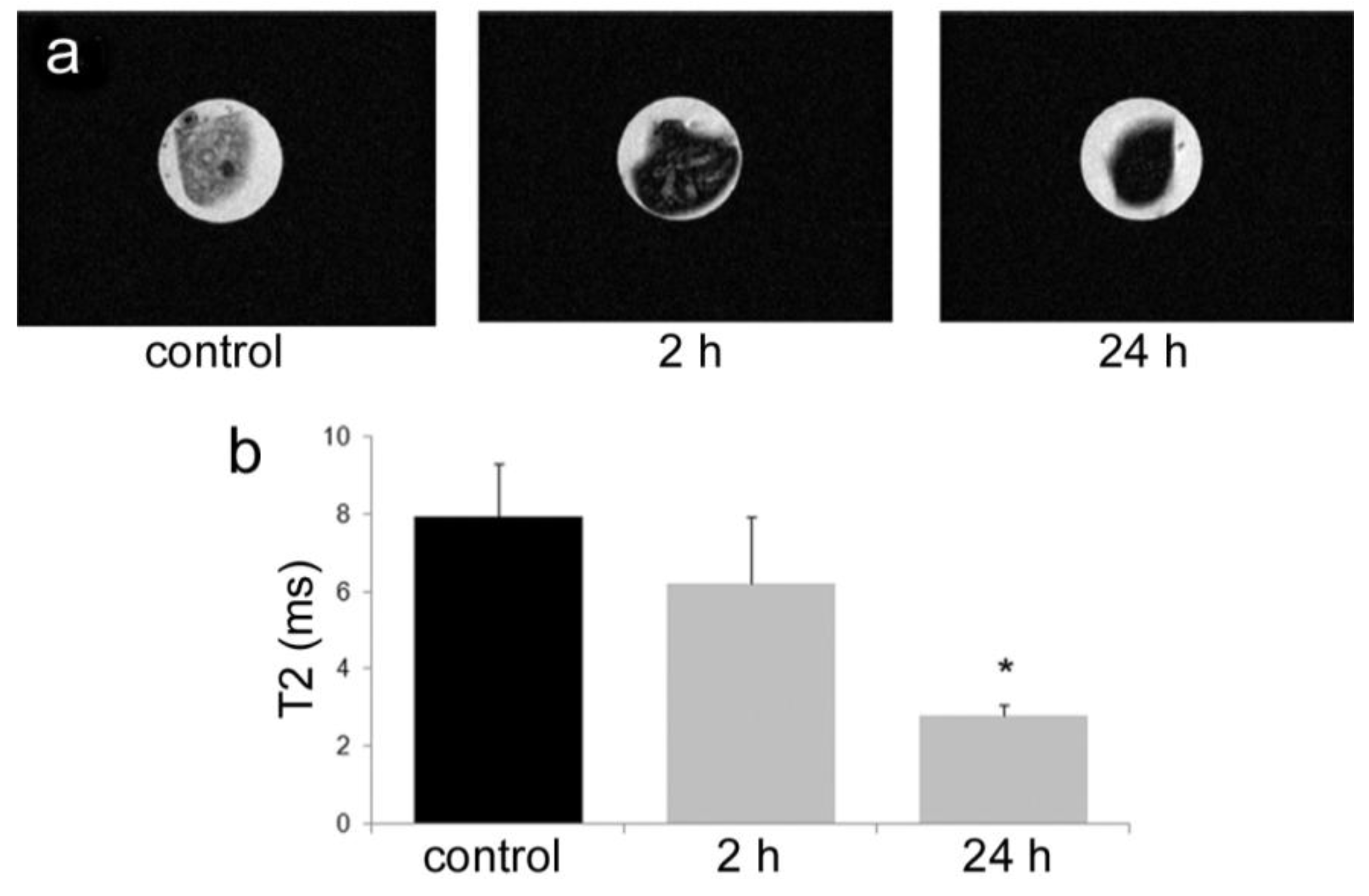
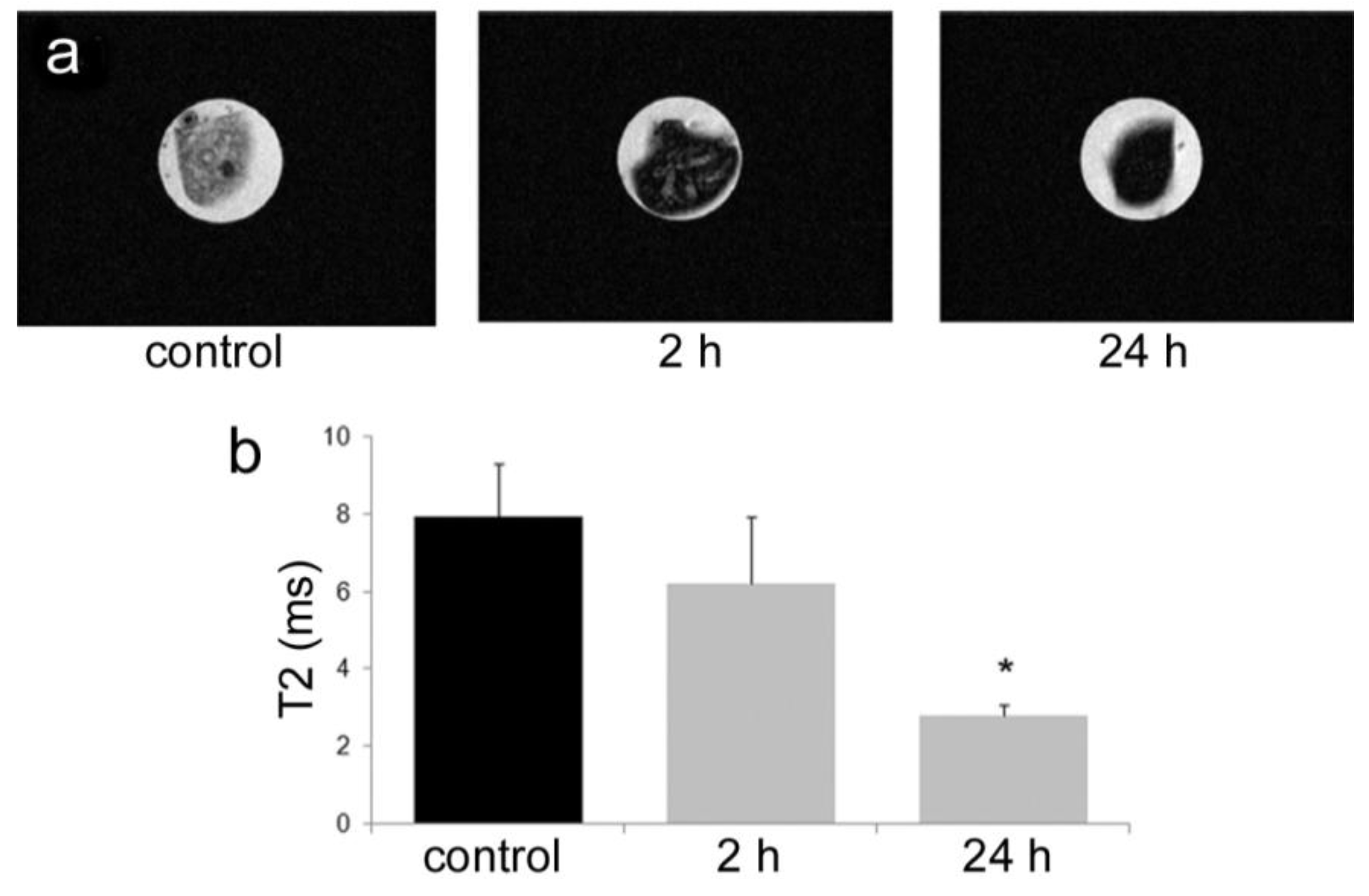
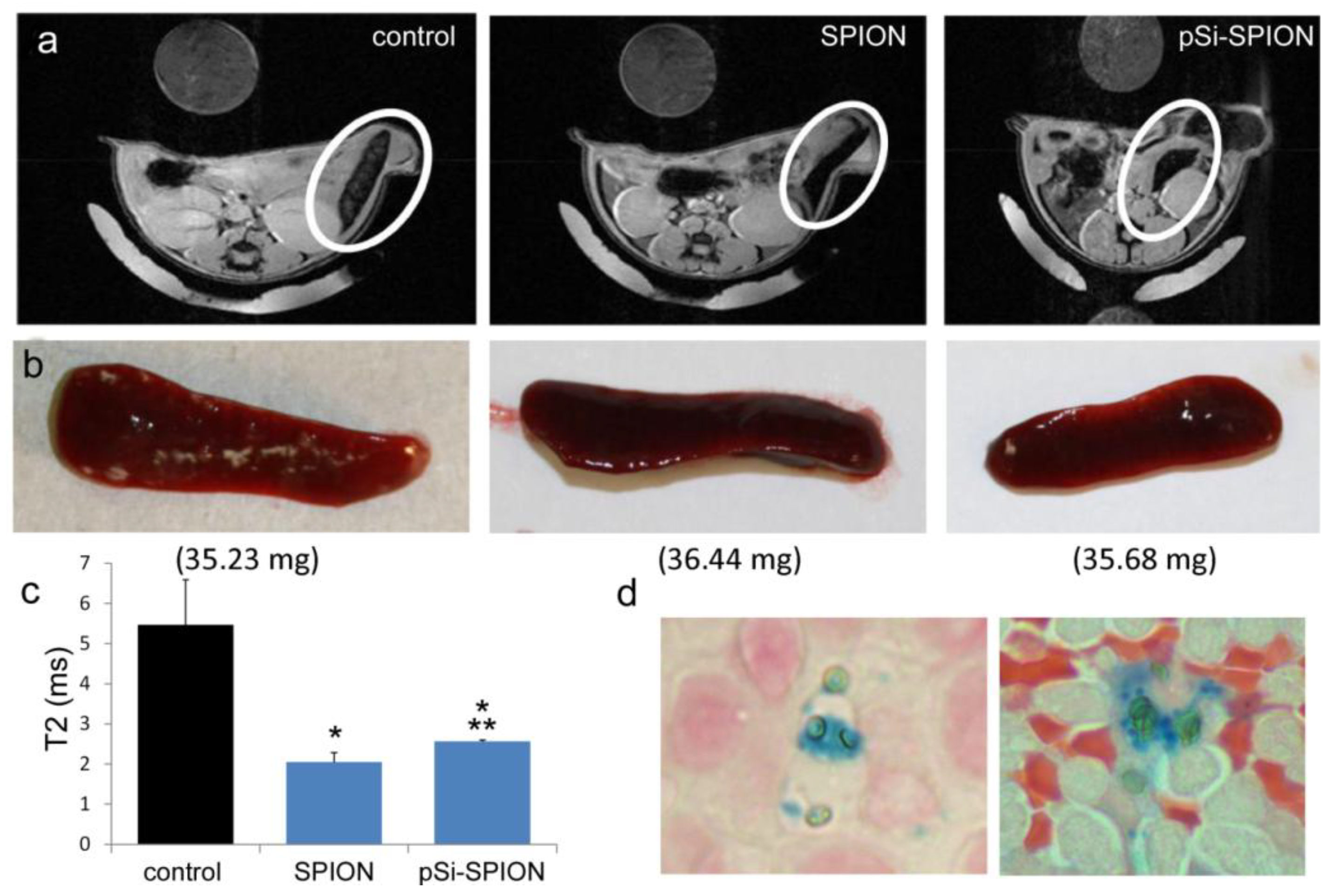
All animal procedures were performed in accordance with recommendations by the National Institutes of Health in the guide “Care and use of Laboratory Animals.” All protocols were reviewed and approved by the Institutional Animal Care and Use Committee at The Methodist Hospital Research Institute (TMHRI), Houston, Texas (Protocol #AUP-1010-0028 and AUP-0311-0012; OLAW Assurance #A4555-01) and at the University of Texas MD Anderson Cancer Center. Time-dependent accumulation of SPIONs in the spleen of female nu/nu mice (Charles River, Wilmington, MA, USA) was determined by intravenous injection of 100 µg SPIONs (Fe content; 20 nm core) in 100 µL PBS. Spleens were excised from mice at 2 and 24 h, suspended in agarose phantoms, and T2-weighted images were acquired using a 7 Tesla scanner as described previously.
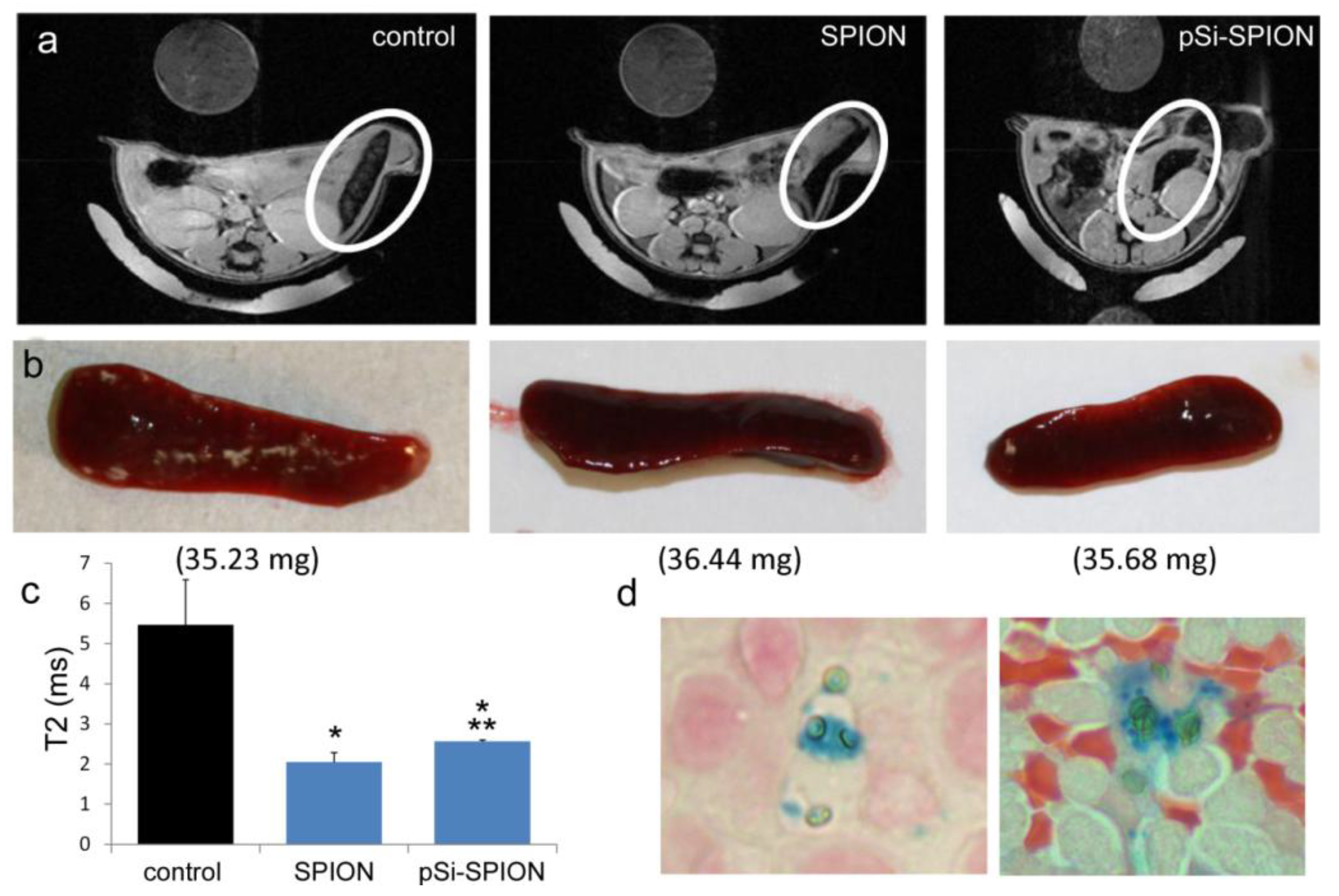
To determine the impact of pSi encapsulation of SPIONs on contrast enhancement in live animals, mice were intravenously injected with PBS or 100 μg of 30 nm SPIONs (free or pSi-encapsulated). T
2-weighted images were taken 24 h post-injection. Signal intensities were determined for 5 regions of interest (mid-section of 5 slices), with normalization against a reference phantom containing 0.01 mg/mL SPIONs or against the psoas muscle. Spleens were isolated and embedded in paraffin 24 h after pSi-SPION injections. Tissue sections were labeled with Prussian blue and Nuclear Fast Red as previously described [
19].
2.9. Preparation of Adjuvant Particles
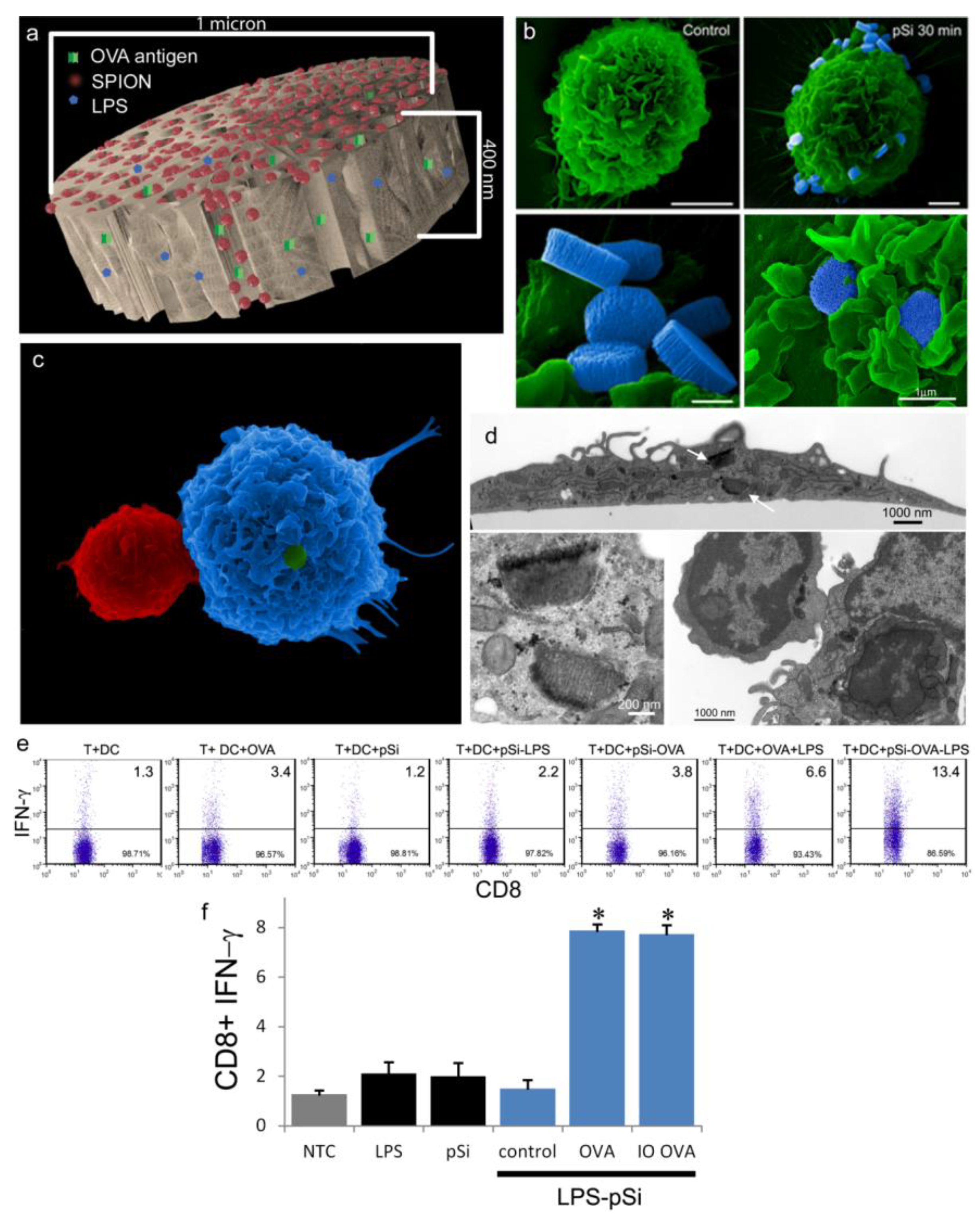
An aqueous 1 mg/mL solution of lipopolysaccharide (LPS) from Escherichia coli (400 µg; Sigma-Aldrich, St. Louis, MO, USA) was treated with ethyl-dimethylaminopropyl carbodiimide (270 µg) to convert carboxyl units to amine-reactive intermediates (final volume 500 µL), and then introduced to APTES-modified pSi microparticles to create stable amide bonds (2 h at 30 °C with agitation at 1400 rpm), followed by washing. LPS-adsorbed (1 mg/mL) microparticles (3 × 108), dried in a desicator, were loaded by capillary action as previously described using a solution made by mixing 50 µL OVA peptide (SIINFEKL (Genscript, Piscataway, NJ, USA); 10 µg/mL) with 50 µL (50 µg) amine-modified SPIONs (20 nm).
2.10. Intracellular Interferon Assay
C57BL/6 (Charles River Laboratories, Inc.; Wilmington, MA, USA) BMDC were prepared by culturing bone marrow cells in granulocyte macrophage colony stimulating factor (20 ng/mL) and interleukin (IL)-4 (100 ng/mL) for 5 days. Splenic T-cells suspensions were prepared from OT-1 mice (The Jackson Laboratory, Bar Harbor, ME, USA) by passing cells through a 70 µm cell strainer and enriching T cells using a Dynal
® Mouse T cell Negative Isolation kit (Invitrogen, Grand Island, NY, USA). BMDC were incubated with microparticles (20 to 1 ratio) for 2 h followed by 18 h co-culture with T cells at a one-to-one ratio (5 × 10
5 each). Cells were permeabilized with Cytofix/Cytoperm™ solution and incubated with PE-conjugated anti-interferon gamma (IFN-γ) and FITC-conjugated anti-CD8 antibodies (BD Biosciences, Mountain View, CA, USA) for 30 min at 4 °C. Samples were analyzed using a LSRFortessa™ Flow Cytometer (BD Biosciences, San Jose, CA, USA) using FACSDIVA™ software (BD Biosciences, San Jose, CA, USA) [
20]. Statistical analysis was based on a two-tailed, equal variance
t-test (
n = 3 per group).
2.11. Electron Microscopy Imaging of Particles and BMDC
SPIONs were suspended in water and dried on a formvar 400 mesh grid. Images were acquired using a JEOL 1200 transmission electron microscope (TEM, JEOL, Peabody, MA, USA) at 60 kV with digital images collected using a 1 k × 1 k Gatan BioScan camera Model 792.
BMDC were cultured on poly-l-lysine glass slides with LPS-bound pSi microparticles (1:20 ratio) in the presence of 10 μg/mL SIINFEKL peptide for 3 h at 37 °C. OT-1 splenic T cells were introduced at a ratio of 2:1 (BMDC:T) and cells were incubated for an additional hour. Following fixation in 2% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, cells were incubated in a mixture of osmium tetroxide (OsO4) and 1% potassium ferrocyanate in 0.1 M cacodylate buffer for 30 min at 48 °C. Cells were then dehydrated and embedded with a mixture of epon and araldite. Ultrathin sections were counter-stained with uranyl acetate and imaged using a JEOL 1210 transmission electron microscope equipped with an AMT Imaging System. Gamma adjustments were made to the micrographs to enhance image contrast and brightness.
For surface topography imaging by scanning electron microscopy (SEM), BMDC were plated in 24 well plates containing 5 × 7 mm silicon chip specimen supports (Ted Pella, Inc., Redding, CA, USA) at 1 × 10
5 cells per well. The next day, cells were incubated with pSi microparticles for 3 h at 37 °C (1:10; cell:microparticle ratio). BMDC were either fixed and dehydrated as previously described [
18,
21] for SEM imaging or further incubated with OT-1 T cells for an additional hour then processed for imaging. Cells were sputter-coated with a 4 nm layer of platinum/palladium (80:20) using a Cressington Sputter Coater 208 HR (Ted Pella, Inc., Redding, CA, USA). SEM images were acquired under high vacuum, at 10 kV, spot size 3.0, using a Nova™ NanoSEM (FEI Company, Hillsboro, OR, USA). Images were pseudo-colored and gamma levels were adjusted to enhance image contrast and brightness.
2.12. Statistical Analysis
Relaxivity data is expressed as the slope of the relaxation rate
vs. concentration. The slope was calculated by best fit linear regression analysis using the least squares method. 95% confidence intervals were generated using the standard error of the slope (SE
slope), given by the following equation:
where
yi is the value of relaxation rate for instance
i, ŷ
i is estimated value of the relaxation rate for observation
i,
xi is the observed value of the concentration for observation
i,
x is the mean of the concentration, and
n is the number of data points (
n = 5). The 95% confidence interval was then calculated using the formula
Regression analysis was done using a single set of relaxation rate measurements vs. concentration for each of the free or encapsulated SPION particle preparations analyzed. Significance was determined with non-overlapping confidence intervals.
4. Discussion
Previously, we demonstrated that loading of SPIONs into pSi microparticles creates hybrid particles with magnetic properties, with increasing payloads of SPIONs inducing greater negative contrast in MR images [
19]. During the loading process, increases in the concentration of SPIONs introduced to dry pSi microparticles increases nanoparticle loading in the silicon pores, with a plateau reached at concentrations above 1 mg/mL [
19] Theoretically, the pore volume of each 1000 × 400 nm pSi microparticle is about 0.2 μm
3, permitting loading of up to 3.8 × 10
5 10 nm nanoparticles. Empirically, we previously demonstrated loading of 0.3 pg of 10 nm SPIONs per pSi microparticle, corresponding to approximately 1 × 10
5 SPIONs [
25].
This manuscript expands on previous work by showing the impact of SPION size and encapsulation in the pores of a silicon microparticle on negative contrast enhancement. As stated previously, relaxation effects of SPIONs are governed by their magnetic properties which are determined by particle size, shape and morphology. As reported by others, we demonstrated that increasing core sizes generates superior relaxivities [
26]. At the level of external magnetic field strength of most clinical scanners (1.5–3 Tesla), the induced magnetization of SPION contrast agents is very close to the asymptotic upper limit, referred to as the induced magnetization at saturation. As the diameter of SPIONs is increased, the induced magnetization at saturation also increases [
27], causing increases in T
2 and T
2* relaxivity [
28]. Plotting relaxation rates (T
2, T
2*) against concentration (mM free iron) generates slopes which yield the concentration-independent relaxivities (r
2 and r
2*). In this study, using a 7 Tesla scanner, we demonstrate that SPION relaxivities increase with particle diameters ranging from 5 to 30 nm, with iron saturation effects becoming apparent for r
2 values for particles 20 nm and higher, and r
2* showing a further increase as the core size increases from 20 to 30 nm.
Accumulation of ultrasmall SPIONs in the phagosomes of macrophages has been shown to enhance decreases in the signal intensity of T
2*-weighted images due to shortening of T
2 and T
2*, with tissue clustering leading to additional T
2* shortening effects [
29]. Similarly, spatial confinement of SPIONs in larger nanoparticles, such as micelles, has been shown to produce strong field gradients leading to increased transverse relaxivities [
30,
31]. Kinsella
et al. [
32] reported that encapsulation of 9 nm Fe
3O
4 in 16 nm silicon pores led to an increase in magnetic strength causing an increase in transverse relaxivity. In this study, encapsulation of 30 nm SPIONs within a pSi matrix (50 nm pores) caused a slight decrease (
p < 0.05) in T
2 transverse relaxation, but no significant impact on T
2* values. There was no increase in the ratio of R
2* to R
2 with compartmentalization of SPIONs in pSi, as predicted by SDR theory. In contrast to previous work by our group [
33] that showed large alterations in T
1 contrast with encapsulation of gadolinium within pSi microparticles, the small enhancement in T
2 relaxivity due to encapsulation of SPIONs in pSi microparticles is not likely to add significant clinical benefit on its own. However, benefits of using hybrid particle complexes, as opposed to free SPIONs, are due to incorporation of additional biologically active agents, such as immune modulators and antigens, achieving delivery of both imaging and therapeutic agents to the same target cells.
As previously demonstrated by others, free SPIONs accumulate predominately in the liver and spleen following intravenous administration [
34], with size and surface modification of the nanoparticles impacting biodistribution [
35]. Time-dependent accumulation of SPIONs in the spleen with time (
Figure 4) was confirmed, with greater levels of SPIONs detected at 24 h compared to 2 h by MRI. Based on higher accumulation at the later time point, 24 h was used for
in vivo MR imaging studies designed to compare accumulation (
i.e., detection) of free verses pSi encapsulated SPIONs. Detection of free SPION accumulation in the spleen was slightly superior to pSi-encapsulated SPIONs following intravascular injection. Future work will examine particle accumulation in the draining lymph nodes following subcutaneous injection to explore the impact of mode of transportation (
i.e., cell-based transport for larger particles verses exit from capillaries and trafficking through the lymphatic system for smaller particles [
17]) on particle accumulation in lymphatic tissue. In addition, we previously demonstrated that particle surface charge alters accumulation of microparticles in the spleen following intravenous administration, with cationic microparticles accumulating preferentially over anionic microparticles [
21]. Since both particle charge and macromolecule binding (e.g., opsonization or ligand conjugation) to the particle surface influence biodistribution, future studies will also examine the impact of loading microparticles with antigens and immune modulators on trafficking of vaccine components to lymphatic tissue.
As demonstrated previously, we show abundant adhesion and uptake of pSi microparticles by BMDC. Loading of antigen and immune modulators (i.e., LPS) stimulates antigen driven interactions between OT-1 T cells and particle-treated BMDC, with enhanced production of IFN-γ in activated CD8+ T cells. Incorporation of SPIONs into the particle complex did not alter stimulation of T cell immunity, supporting the use of the hybrid particles for the development of immunotherapeutics.

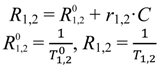
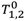 is the relaxation time of the medium with contrast agent removed, C is the concentration of contrast agent, and r1,2 is the concentration-independent relaxivity of the contrast agent under study. The term R1,2 in the above equation is referred to as the relaxation rate.
is the relaxation time of the medium with contrast agent removed, C is the concentration of contrast agent, and r1,2 is the concentration-independent relaxivity of the contrast agent under study. The term R1,2 in the above equation is referred to as the relaxation rate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}