Acute Traumatic Endotheliopathy in Isolated Severe Brain Injury and Its Impact on Clinical Outcome

 ,
,
Abstract
:1. Introduction
1.1. Background
1.2. Objectives
2. Materials and Methods
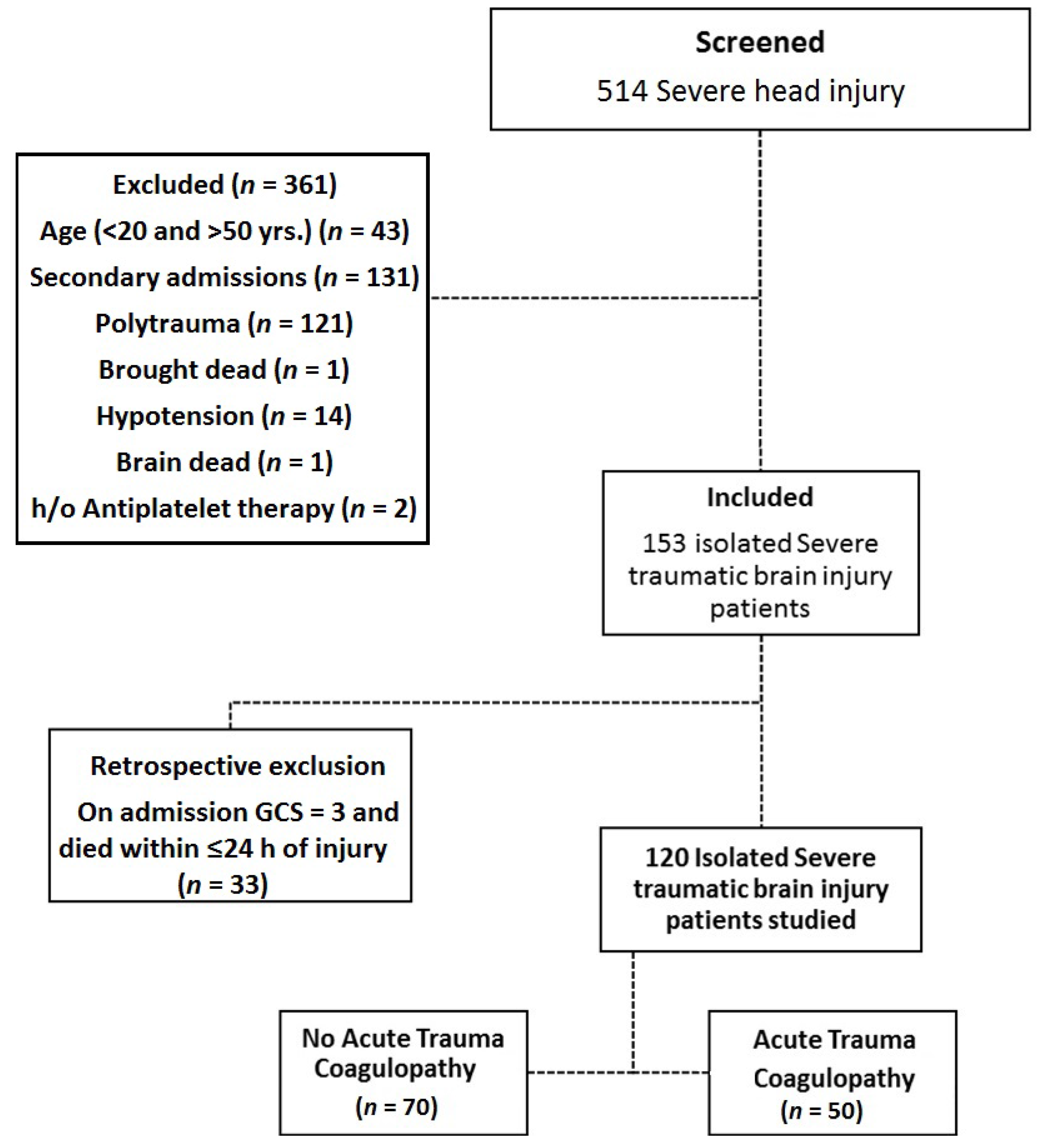
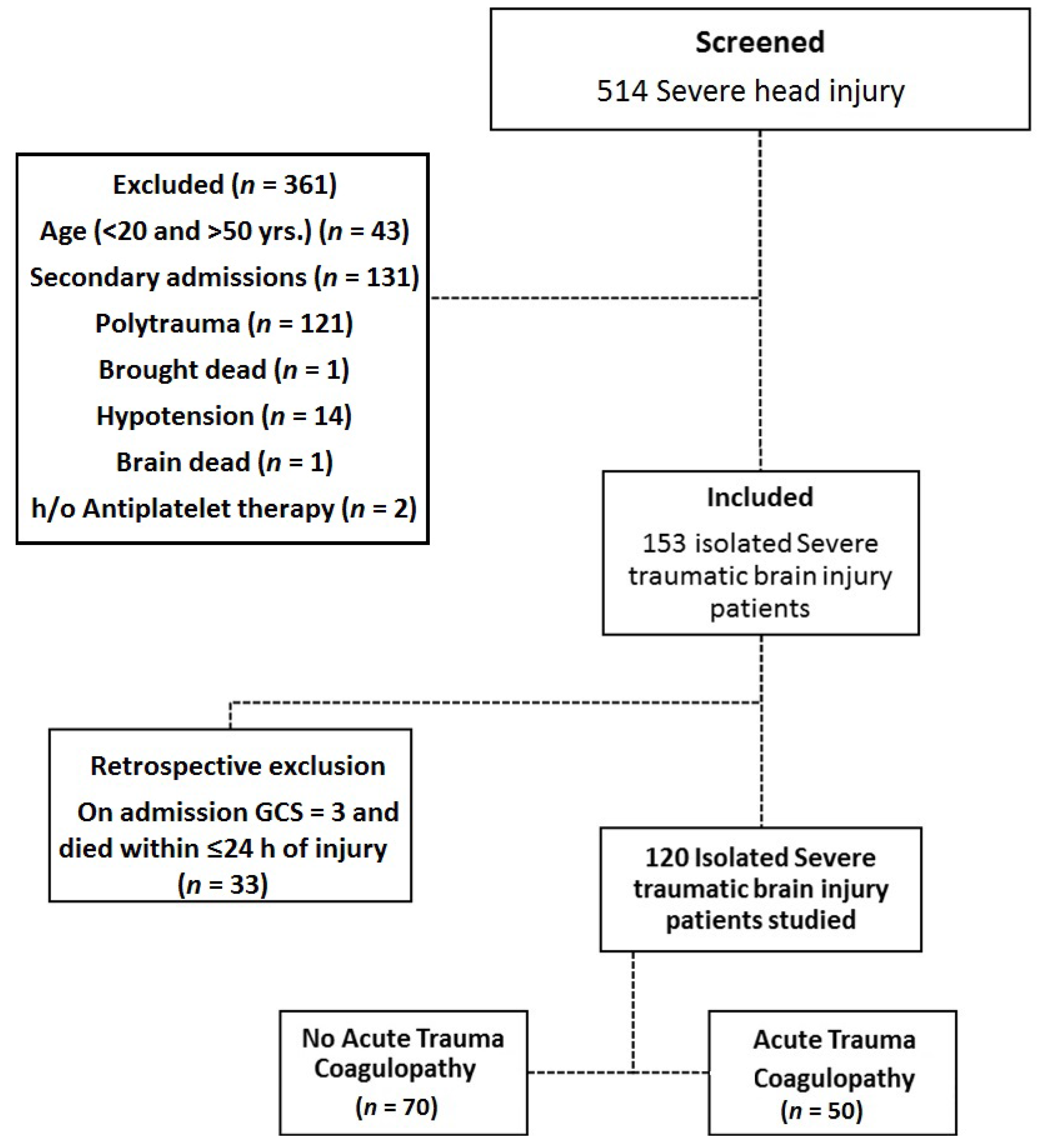
2.1. Study Design, Participants and Setting
2.2. Defining Isolated TBI
2.3. Defining TBI Associated Coagulopathy
2.4. Sample Size
2.5. Sample Processing and Analysis
2.5.1. Blood
2.5.2. Enzyme Linked Immunosorbent assay
2.6. Study Variables
2.7. Statistical Methods
3. Results
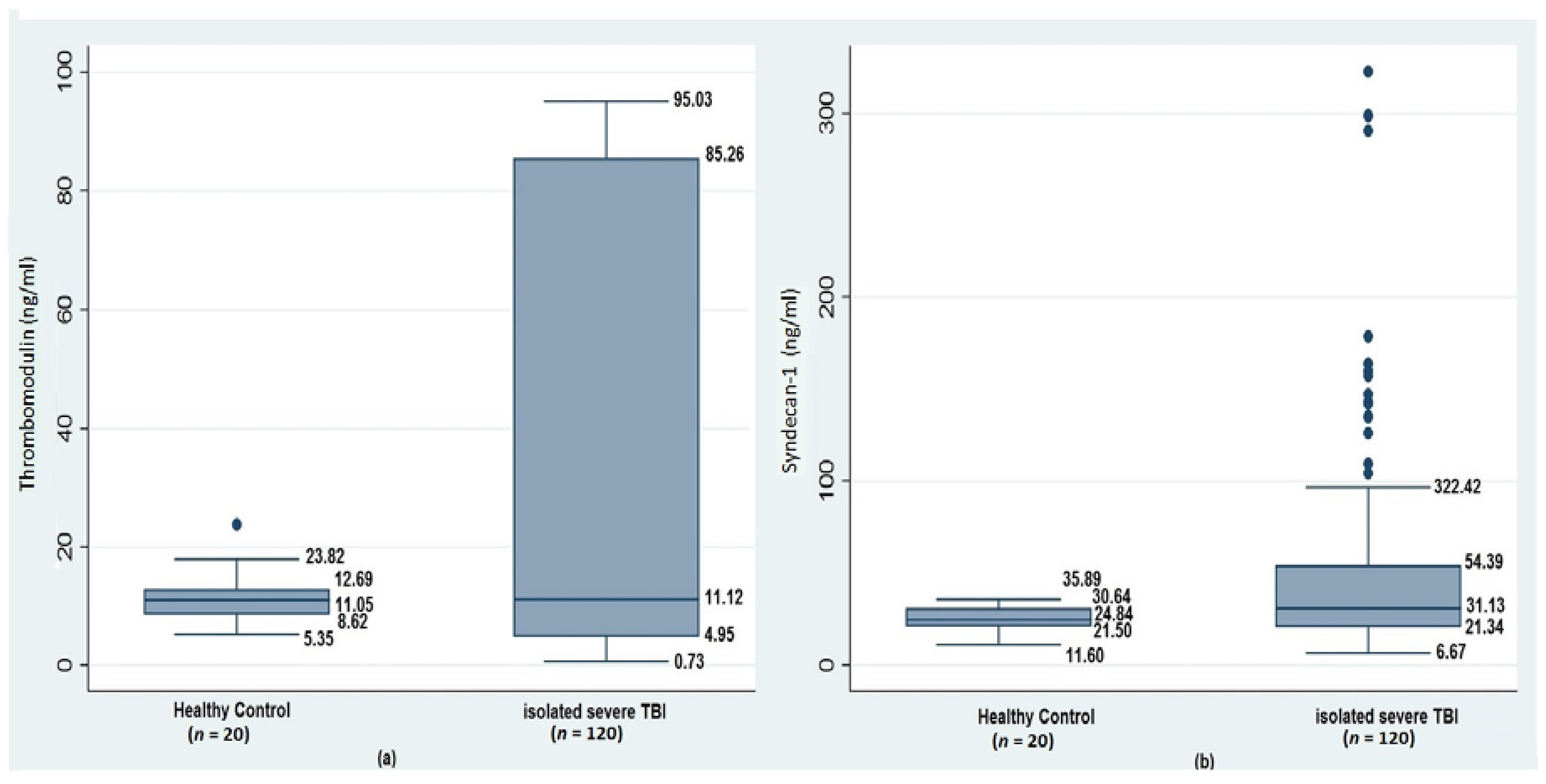
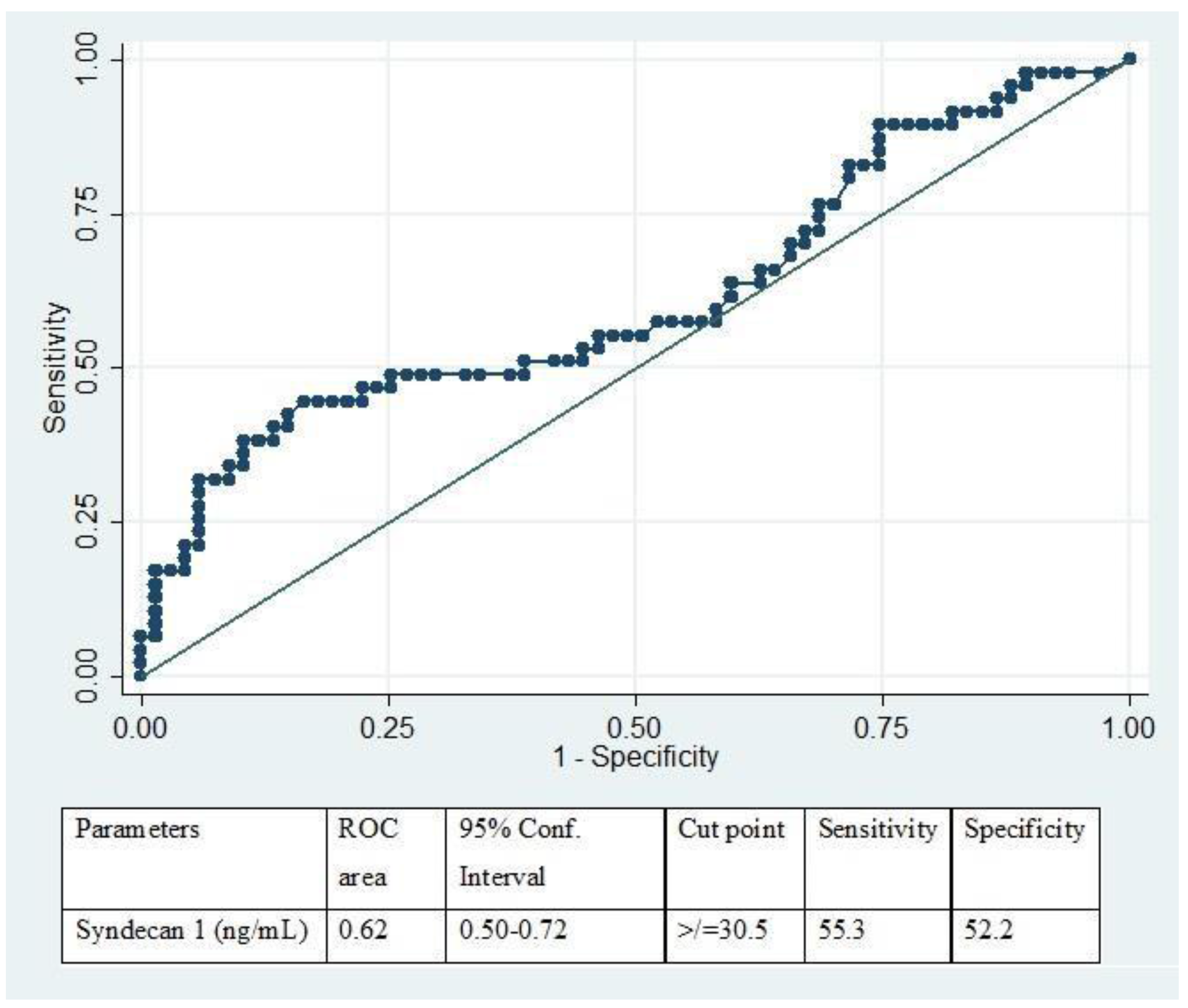
3.1. Acute Coagulopathy, Glycocalyx Shedding, and Endothelial Disruption of Traumatic Brain Injury
3.2. Clinical Outcome
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kauvar, D.S.; Lefering, R.; Wade, C.E. Impact of haemorrhage on trauma outcome: An overview of epidemiology, clinical presentations, and therapeutic considerations. J. Trauma Acute Care Surg. 2006, 60, S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Oertel, M.; Kelly, D.F.; McArthur, D.; Boscardin, W.J.; Glenn, T.C.; Lee, J.H.; Tooraj Gravori, M.P.H.; Obukhov, D.; McBride, D.Q.; Martin, N.A. Progressive hemorrhage after head trauma: Predictors and consequences of the evolving injury. J. Neurosurg. 2002, 96, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Harhangi, B.S.; Kompanje, E.J.; Leebeek, F.W.; Maas, A.I. Coagulation disorders after traumatic brain injury. Acta Neurochir. 2008, 150, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, R.; Liu, L.; Watkins, T.; Zhang, F.; Dong, J. Traumatic Brain Injury-Associated Coagulopathy. J. Neurotrauma 2012, 29, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Epstein, D.S.; Mitra, B.; O’Reilly, G.; Rosenfeld, J.V.; Cameron, P.A. Acute traumatic coagulopathy in the setting of isolated traumatic brain injury: A systematic review and metaanalysis. Injury 2014, 45, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalik, P.; Boorman, D.; Tracy, J.; Jallo, J.; Rincon, F. Acute traumatic coagulopathy accompanying isolated traumatic brain injury is associated with worse long-term functional and cognitive outcomes. Neurocrit. Care 2016, 24, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Maegele, M. Coagulopathy after traumatic brain injury: Incidence, pathogenesis, and treatment options. Transfusion 2013, 53, 28S–37S. [Google Scholar] [CrossRef] [PubMed]
- Hemker, H.C.; Dieri Al, R.; Béguin, S. Thrombin generation assays: Accruing clinical relevance. Curr. Opin. Hematol. 2004, 11, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Martini, W.Z.; Dubick, M.A.; Salinas, J.; Butenas, S.; Kheirabadi, B.S.; Pusateri, A.E.; Vos, J.A.; Guymon, C.H.; Wolf, S.E.; et al. Thromboelastography as a better indicator of hypercoagulable state after injury than prothrombin time or activated partial thromboplastin time. J. Trauma 2009, 67, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Dobson, G.P.; Letson, H.L.; Sharma, R.; Sheppard, F.R.; Cap, A.P. Mechanisms of early trauma-induced coagulopathy: The clot thickens or not? J. Trauma Acute Care Surg. 2015, 79, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Cap, A.P.; Spinella, P.C. Severity of head injury is associated with increased risk of coagulopathy in combat casualties. J. Trauma Acute Care Surg. 2011, 71, S78–S81. [Google Scholar] [CrossRef] [PubMed]
- Genét, G.F.; Johansson, P.I.I.; Meyer, M.A.; Sølbeck, S.; Sørensen, A.M.; Larsen, C.F.; Welling, K.L.; Windeløv, N.A.; Rasmussen, L.S.; Ostrowski, S.R. Trauma-induced coagulopathy: Standard coagulation tests, biomarkers of coagulopathy, and endothelial damage in patients with traumatic brain injury. J. Neurotrauma 2013, 30, 301–306. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Manoel, A.L.; Neto, A.C.; Veigas, P.V.; Rizoli, S. Traumatic brain injury associated coagulopathy. Neurocrit. Care 2014, 22, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Matthay, M.A.; Mackersie, R.C.; Pittet, J.-F. Acute traumatic coagulopathy: Initiated by hypoperfusion: Modulated through the protein C pathway? Ann. Surg. 2007, 245, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Schultz, M.J.; Levi, M.; Mackersie, R.C.; Pittet, J.-F. Acute coagulopathy of trauma: Hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J. Trauma Acute Care Surg. 2008, 64, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Johansson, P.I. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J. Trauma Acute Care Surg. 2012, 73, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Haase, N.; Müller, R.B.; Møller, M.H.; Pott, F.C.; Perner, A.; Johansson, P.I. Association between biomarkers of endothelial injury and hypocoagulability in patients with severe sepsis: A prospective study. Crit. Care 2015, 19, 191. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Haase, N.; Perner, A.; Ostrowski, S.R. Association between sympathoadrenal activation, fibrinolysis, and endothelial damage in septic patients: A prospective study. J. Crit. Care 2014, 29, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W. Endothelium—Role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Chernow, B.; Rainey, T.G.; Lake, C.R. Endogenous and exogenous catecholaminesin critical care medicine. Crit. Care Med. 1982, 10, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Von Kanel, R.; Dimsdale, J.E. Effects of sympathetic activation by adrenergicinfusions on hemostasis in vivo. Eur. J. Haematol. 2000, 65, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann. Surg. 2011, 254, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Danielli, J.F. Capillary permeability and oedema in the perfused frog. J. Physiol. 1940, 98, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.F.; Chappell, D.; Bruegger, D.; Annecke, T.; Jacob, M. Therapeutic strategies targeting the endothelial glycocalyx: Acute deficits, but great potential. Cardiovasc. Res. 2010, 87, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, T.E.; Woodcock, T.M. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: An improved paradigm for prescribing intravenous fluid therapy. Br. J. Anaesth. 2012, 108, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.E.; Johansson, P.I.; Ostrowski, S.R.; Bestle, M.H.; Hein, L.; Jensen, A.L.; Søe-Jensen, P.; Andersen, M.H.; Steensen, M.; Mohr, T.; et al. Profound endothelial damage predicts impending organ failure and death in sepsis. Semin. Thromb. Hemost. 2015, 41, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Schött, U.; Solomon, C.; Fries, D.; Bentzer, P. The endothelial glycocalyx and its disruption, protection and regeneration: A narrative review. Scand. J. Trauma Resusc. Emerg. Med. 2016, 24, 48. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, E.; Cardenas, J.C.; Baimukanova, G.; Usadi, B.; Bruhn, R.; Pati, S. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J. Transl. Med. 2015, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. High circulating adrenaline levels at admission predict increased mortality after trauma. J. Trauma Acute Care Surg. 2012, 72, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Haywood-Watson, R.J.; Holcomb, J.B.; Gonzalez, E.A.; Peng, Z.; Pati, S.; Park, P.W.; Wang, W.; Zaske, A.M.; Menge, T.; Kozar, R.A. Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS ONE 2011, 6, e23530. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Ostrowski, S.R. Acute coagulopathy of trauma: Balancing progressive catecholamine induced endothelial activation and damage by fluid phase anticoagulation. Med. Hypotheses 2010, 75, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Nieuwdorp, M.; van Haeften, T.W.; Gouverneur, M.C.; Mooij, H.L.; van Lieshout, M.H.; Levi, M.; Meijers, J.C.M.; Holleman, F.; Hoekstra, J.B.L.; Vink, H.; et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 2006, 55, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I.R. Position statement: Definition of traumatic brain injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Greuters, S.; van den Berg, A.; Franschman, G.; Viersen, V.A.; Beishuizen, A.; Peerdeman, S.M.; Boer, C. Acute and delayed mild coagulopathy are related to outcome in patients with isolated traumatic brain injury. Crit. Care 2011, 15, R2. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Rodriguez, E.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Tomasek, J.S. Syndecan-1: A Quantitative Marker for the Endotheliopathy of Trauma. J. Am. Coll. Surg. 2017, 225, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Sillesen, M.; Rasmussen, L.S.; Jin, G.; Jepsen, C.H.; Imam, A.; Hwabejire, J.O.; Halaweish, I.; DeMoya, M.; Velmahos, G.; Johansson, P.I.; et al. Assessment of coagulopathy, endothelial injury, and inflammation after traumatic brain injury and hemorrhage in a porcine model. J. Trauma Acute Care Surg. 2014, 76, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.P.; Moore, E.E.; Moore, H.B. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J. Trauma Acute Care Surg. 2015, 80, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Yokota, H.; Naoe, Y.; Nakabayashi, M.; Unemoto, K.; Kushimoto, S.; Kurokawa, A.; Node, Y.; Yamamoto, Y. Cerebral endothelial injury in severe head injury: The significance of measurements of serum thrombomodulin and the von Willebrand factor. J. Neurotrauma 2002, 19, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Rehm, M.; Bruegger, D.; Christ, F.; Conzen, P.; Thiel, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Reichart, B.; et al. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation 2007, 116, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, J.B. A novel and potentially unifying mechanism for shock induced early coagulopathy. Ann. Surg. 2011, 254, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Di Battista, A.P.; Rizoli, S.B.; Lejnieks, B.; Min, A.; Shiu, M.Y.; Peng, H.T.; Baker, A.J.; Hutchison, M.G.; Churchill, N.; Inaba, K.; et al. Sympathoadrenal activation is associated with acute traumatic coagulopathy and endotheliopathy in isolated brain injury. Shock 2016, 46, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Chignalia, A.Z.; Yetimakman, F.; Christiaans, S.C.; Unal, S.; Bayrakci, B.; Wagener, B.M.; Russell, R.T.; Kerby, J.D.; Pittet, J.-F.F.; Dull, R.O. The glycocalyx and trauma: A review. Shock 2016, 45, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A. Sympathoadrenal activation and endotheliopathy are drivers of hypocoagulability and hyperfibrinolysis in trauma: A prospective observational study of 404 severely injured patients. J. Trauma Acute Care Surg. 2017, 82, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Iseki, K.; Hagino, S.; Mori, T.; Zhang, Y.; Yokoya, S.; Takaki, H.; Tase, C.; Murakawa, M.; Wanaka, A. Increased syndecan expression by pleiotrophin and FGF receptor-expressing astrocytes in injured brain tissue. Glia 2002, 39, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, J.B.; Pati, S. Optimal Trauma Resuscitation with Plasma as the Primary Resuscitative Fluid: The Surgeon’s Perspective; ASH Education Program Book; American Society of Hematology: Washington, DC, USA, 2013; pp. 656–659. [Google Scholar]
- Puskarich, M.A.; Cornelius, D.C.; Tharp, J.; Nandi, U.; Jones, A.E. Plasma syndecan-1 levels identify a cohort of patients with severe sepsis at high risk for intubation after large-volume intravenous fluid resuscitation. J. Crit. Care 2016, 36, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Naumann, D.N.; Hazeldine, J.; Davies, D.J.; Bishop, J.; Midwinter, M.J.; Belli, A.; Harrison, P.; Lord, J.M. Endotheliopathy of Trauma is an On-Scene Phenomenon, and is Associated with Multiple Organ Dysfunction Syndrome: A Prospective Observational Study. Shock 2017. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, J.; Wu, X.; Xi, C.; Gai, Y.; Liu, H.; Yuan, Q.; Wang, E.; Gao, L.; Hu, J.; et al. Validating the incidence of coagulopathy and disseminated intravascular coagulation in patients with traumatic brain injury—Analysis of 242 cases. Br. J. Neurosurg. 2011, 25, 363–368. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | Overall (n = 120) | Study Group (n = 120) | No TBI-AC vs. TBI-AC p-Value | ||
|---|---|---|---|---|---|
| No TBI-AC (n = 70) | TBI-AC (n = 50) | ||||
| Age * (years) | 35.5 ± 12.6 | 36.0 ± 12.89 | 34.8 ± 13.35 | 0.62 | |
| Gender † | Male | 106(88.3) | 61(87.1) | 45(90.0) | 0.63 |
| Female | 14(11.6) | 9(12.9) | 5(10.0) | ||
| Mode of injury † | RTA | 83(69.2) | 48(68.5) | 35(70.0) | 0.83 |
| Fall | 21(17.5) | 12(17.1) | 9(18.0) | ||
| Assault | 2(1.67) | 2(2.8) | 0 | ||
| Miscellaneous | 14(11.7) | 8(11.4) | 6(12.0) | ||
| Time taken from injury to admission * (h) | 5(1–5) | 2(1–5) | 2(1–4) | 0.71 | |
| Mechanical ventilation † | No | 44(36.7) | 23(32.8) | 21(42.0) | 0.30 |
| Yes | 76(63.3) | 47(67.2) | 29(58.0) | ||
| Systolic BP* (mm/Hg) | 139.5 ± 26.4 | 143.1 ± 3.2 | 134.3 ± 3.4 | 0.07 | |
| Glasgow Coma Scale | 7(5–8) | 7(5–8) | 6(5–7) | 0.09 | |
| Hypoperfusion † (n = 72) | No | 52(72.2) | 33(78.5) | 19(63.3) | 0.15 |
| Yes | 20(27.8) | 9(21.6) | 11(36.4) | ||
| Acidosis † (n = 68) | No | 39(57.4) | 28(70.0) | 11(39.2) | 0.01 |
| Yes | 29(42.6) | 12(30.0) | 17(60.3) | ||
| Haemoglobin (g/dL) | 12.5 ± 2.69 | 12.7 ± 2.50 | 12.3 ± 2.94 | 0.35 | |
| Anaemia † (Hb < 9gm/dL) | No | 104(86.7) | 61(87.1) | 43(86.0) | 0.85 |
| Yes | 16(13.3) | 9(12.9) | 7(14.0) | ||
| Red blood cell count (106 cumm) | 4.3 ± 0.85 | 4.4 ± 0.78 | 4.1 ± 0.93 | 0.06 | |
| Haematocrit (%) | 39.7 ± 7.82 | 40.4 ± 7.13 | 38.6 ± 8.67 | 0.20 | |
| Platelet count * (103/cumm) | 193(134.5–250.5) | 196.5(138.0–248.0) | 184.5(116.0–251.0) | 0.53 | |
| Thrombocytopenia † (plt < 100 × 103 cumm) | No | 105(87.5) | 63(90) | 42(84) | 0.32 |
| Yes | 15(12.5) | 7(10) | 8(16) | ||
| White blood cell count * (4.0–11.0 × 103/cumm) | 14.3(10.8–18.7) | 13.9(11.0–19.1) | 14.7(10.8–18.4) | 0.79 | |
| Prothrombin time (s) | 16.0 ± 6.1 | 13.9 ± 1.1 | 18.8 ± 8.51 | <0.0001 | |
| activated partial thromboplastin time (aPTT) (s) | 29.6 ± 13.7 | 24.1 ± 2.4 | 37.3 ± 18.5 | <0.0001 | |
| International normalized ratio (INR) | 1.0 ± 0.36 | 1.0 ± 0.12 | 1.4 ± 0.46 | <0.0001 | |
| Hospital length of stay † (days) | 8(4–20) | 7(2–16) | 9(5–21) | 0.73 | |
| ICU length of stay † (days) | 5(3–9) | 5(3–8) | 5(2–9) | 0.91 | |
| Transfusion requirements † | PRBC (n = 57) | 0(0–4) | 0(0–3) | 2(0–6) | 1.0 |
| FFP (n = 34) | 0(0–2.5) | 0(0–0) | 0(0–4) | ||
| PRP (n = 25) | 0(0–0) | 0(0–0) | 0(0–4) | ||
| Mortality † | No | 84(70) | 56(80.0) | 28(56.0) | 0.005 |
| Yes | 36(30) | 14(20.0) | 22(44.0) | ||
| Parameter | HC (n = 20) | Study Group (n = 120) | p-Value | Post hoc p-value | |
|---|---|---|---|---|---|
| No TBI-AC (n = 70) | TBI-AC (n = 50) | ||||
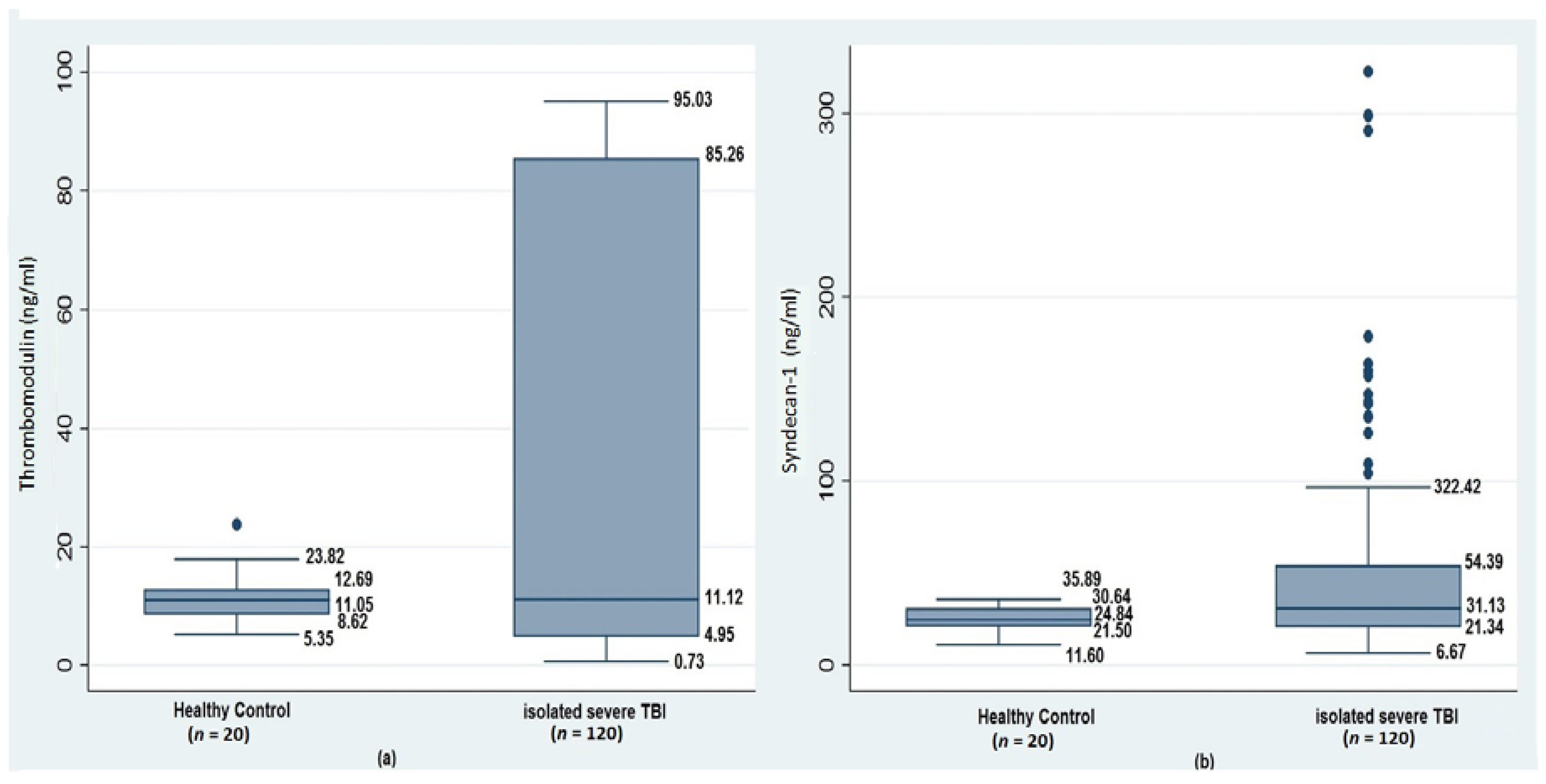
| Thrombomodulin (ng/mL) | 11.0(8.6–12.6) | 11.8(4.9–86.2) | 10.2(3.9–84.0) | 0.76 | - |
| Syndecan-1 (ng/mL) | 24.8(21.5–30.6) | 29.9(19.2–39.5) | 33.7(21.6–109.5) | 0.03 | HC vs. No TBI-AC 0.20 HC vs. TBI-AC 0.04 No TBI-AC vs. TBI-AC 0.03 |
| Clinical Parameters | No TBI-AC (n = 70) | p-Value | TBI-AC (n = 50) | p-Value | ||
|---|---|---|---|---|---|---|
| No-Endotheliopathy (n = 38) | Endotheliopathy (n = 35) | No-Endotheliopathy (n = 21) | Endotheliopathy (n = 26) | |||
| Thrombomodulin (ng/mL) | 11.6(5.6–86.6) | 10.8(3.9–21.7) | 0.65 | 5.5(1.6–10.6) | 20.2(8.0–88.0) | 0.002 |
| Clinical Parameters | No endotheliopathy (n = 61) | Endotheliopathy (n = 59) | p-Value | |
|---|---|---|---|---|
| ICU length of stay (days) | 6(3–11) | 5(2–7) | 0.26 | |
| Hospital length of stay (days) | 8(4–17) | 7(4–20) | 0.49 | |
| PRBC (n = 57) | No | 40(65.6) | 21(35.6) | 0.001 |
| Yes | 21(34.4) | 38(64.4) | ||
| FFP (n = 34) | No | 53(86.9) | 33(55.9) | <0.0001 |
| Yes | 8(13.1) | 26(44.1) | ||
| PRC (n = 25) | No | 54(88.5) | 41(69.5) | 0.01 |
| Yes | 7(11.5) | 18(30.5) | ||
| Sepsis | No | 52(85.3) | 49(83.1) | 0.74 |
| Yes | 9(14.8) | 10(16.9) | ||
| 48-h mortality | No | 57(93.4) | 47(79.6) | 0.02 |
| Yes | 4(6.6) | 12(20.4) | ||
| 30-day mortality | No | 51(83.6) | 33(55.9) | 0.001 |
| Yes | 10(16.4) | 26(44.1) | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albert, V.; Subramanian, A.; Agrawal, D.; Pati, H.P.; Gupta, S.D.; Mukhopadhyay, A.K. Acute Traumatic Endotheliopathy in Isolated Severe Brain Injury and Its Impact on Clinical Outcome. Med. Sci. 2018, 6, 5. https://doi.org/10.3390/medsci6010005
Albert V, Subramanian A, Agrawal D, Pati HP, Gupta SD, Mukhopadhyay AK. Acute Traumatic Endotheliopathy in Isolated Severe Brain Injury and Its Impact on Clinical Outcome. Medical Sciences. 2018; 6(1):5. https://doi.org/10.3390/medsci6010005
Chicago/Turabian StyleAlbert, Venencia, Arulselvi Subramanian, Deepak Agrawal, Hara Prasad Pati, Siddhartha Datta Gupta, and Asok Kumar Mukhopadhyay. 2018. "Acute Traumatic Endotheliopathy in Isolated Severe Brain Injury and Its Impact on Clinical Outcome" Medical Sciences 6, no. 1: 5. https://doi.org/10.3390/medsci6010005
APA StyleAlbert, V., Subramanian, A., Agrawal, D., Pati, H. P., Gupta, S. D., & Mukhopadhyay, A. K. (2018). Acute Traumatic Endotheliopathy in Isolated Severe Brain Injury and Its Impact on Clinical Outcome. Medical Sciences, 6(1), 5. https://doi.org/10.3390/medsci6010005