Real-Time Multidepth Multiphoton Microscopy Using Pixel-to-Pixel Focus-Switching

1
Center for Advanced Laser Technology, Hebei University of Technology, Tianjin 300401, China
2
National Key Laboratory of Science and Technology on Tunable Laser, Harbin Institute of Technology, Harbin 150080, China
*
Author to whom correspondence should be addressed.
Appl. Sci. 2020, 10(20), 7173; https://doi.org/10.3390/app10207173
Submission received: 22 September 2020
/
Revised: 10 October 2020
/
Accepted: 13 October 2020
/
Published: 15 October 2020
(This article belongs to the Special Issue Ultrafast Optics: Trends and Prospects)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Multiphoton microscopy is a well-established technique for biomedical applications, but real-time multidepth multimodal multiphoton microscopy using non-imaging detection has barely been discussed. We demonstrate a novel label-free imaging system capable of generating multimodal multiphoton signals at different focal planes simultaneously. Two spatially overlapped and temporally interlaced beams are obtained by applying cost-effective electro-optic modulator (EOM)-based fast-switching light paths. The switching beams have different divergence properties, enabling imaging at different depths into samples. The EOM is synchronized to the pixel clock from the microscope, achieving pixel-to-pixel focus-switching. The capability of the imaging system is demonstrated by performing real-time multidepth two-photon fluorescence (TPF) and second-harmonic generation (SHG) imaging of freshly excised mouse lung lobes. TPF and SHG images are acquired at two wavelength ranges. One is between 415 and 455 nm, and the other is between 495 and 635 nm. The microenvironment of pulmonary alveoli is depicted by the distributions of both elastin fibers visualized by TPF and collagen fibers illustrated by SHG. Macrophages residing inside apparent alveolar lumens are also identified by TPF, which shows that the imaging system is capable of localizing biological objects in three dimensions and has the potential of monitoring in vivo cellular dynamics in the axial direction.
1. Introduction
Nonlinear optical microscopy has been proven to be a well-established technique based on nonlinear interactions between light and matter [1,2,3,4]. In a nonlinear optical process, the emission intensity has a quadratic or high-order dependence on the excitation intensity, which ensures that the signal collected out of the focal volume reaches its minimum. Thus, nonlinear optical microscopy has submicron three-dimensional spatial resolution with higher signal-to-noise ratio and less out-of-focus photobleaching, compared with traditional confocal microscopy [5,6,7,8,9]. In addition, using near infrared (NIR) excitation wavelengths also increases the penetration depth, which is particularly significant for biomedical imaging [10,11,12]. Multiphoton microscopy (MPM) and coherent Raman scattering (CRS) microscopy are the most frequently applied nonlinear imaging technologies for biomedical microscopy [13,14,15,16,17,18]. The former mainly includes two-photon fluorescence (TPF), three-photon fluorescence (ThPF), second-harmonic generation (SHG), and third-harmonic generation (THG) microscopy. The latter mainly consists of coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS) microscopy.
TPF microscopy has been the most preferred nonlinear microscopy for biological imaging since 1990 [13], thanks to its simpler setup compared to CRS microscopy and its superior ability to image a wider range of specimens compared to other multiphoton microscopy techniques. Intrinsic fluorescent biomolecules in biological tissues can be applied for imaging without the need of exogenous biomarkers, which ensures that TPF microscopy is a label-free technique. These biomolecules, called endogenous fluorophores, exist at both intracellular scale and in the extracellular matrix of biological tissues. The most widely used intracellular fluorophores are metabolic coenzymes, which include reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD). Investigations of physiological processes, e.g., cell differentiation and cancer development, have been performed by using autofluorescence derived from NADH and FAD [19]. Concerning the extracellular matrix, visualization of autofluorescent proteins, e.g., elastin, collagen, and keratin, has been used to diagnose diseases [20] such as asthma and pulmonary fibrosis.
Even though TPF is the most widely used nonlinear process for biological microscopy, the first nonlinear label-free microscope is based on SHG [11]. Photons are scattered by molecules in the process of SHG, rather than absorbed in the process of TPF. SHG emitters are required to be spatially organized in non-centrosymmetric structures to avoid cancellation. In biological materials (e.g., collagen and myosin) with such structures, the signal of SHG could be very intense. The structures of collagen containing tissues, such as skin, tendon, and lung, have been revealed by SHG microscopy [20]. Meanwhile, pathological processes, such as liver fibrosis and hypertrophic scar development, have been investigated by SHG imaging [21].
TPF and SHG collect physiological and structural information from biological samples, respectively. Combining the two modalities will obtain more informative and comprehensive images. Simultaneous label-free TPF and SHG microscopy has been developed based on this concept over the last decade. Several groups have demonstrated simultaneous label-free acquisition of TPF from NADH and SHG from collagen using different excitation wavelengths [19,20,21]. Although these platforms provide high-quality images with sufficient biological information, only one depth can be imaged at the same time. Multifocal-plane microscopy and point-spread function (PSF) engineering are two typical methods to image fluorescent particles at different focal planes simultaneously, and several implementations have been established [22,23,24,25]. However, these systems either require multiple detectors for one imaging modality or are unable to collect signal from scattering specimens. Moreover, it is problematic for these systems to obtain simultaneous TPF and SHG images effectively.
Recently, we demonstrated high-speed TPF microscopy systems based on two-dimensional laser scanning [26,27]. In this paper, we demonstrate a label-free imaging system capable of generating multimodal multiphoton signals at different focal planes simultaneously, by using pixel-to-pixel focus-switching. The major advantages of our imaging system are as follows.
- Real-time multidepth microscopy using non-imaging detection enables localization of biological objects in three dimensions within scattering specimens.
- As the key technique of the imaging system, the electro-optic modulator (EOM)-based fast-switching light paths are cost-effective and can be easily added to any commercial multiphoton microscope without internal modification.
- Simultaneous label-free TPF and SHG imaging can gain functional and structural information together from unstained samples and eliminate possible artifacts from sample motion or laser power fluctuation.
We demonstrate the capability of the imaging system by performing real-time multidepth multiphoton imaging of freshly excised mouse lung lobes at 870 nm excitation. Signals are collected by detection channels at two different wavelength ranges. One is between 415 and 455 nm, and the other is between 495 and 635 nm. Multiphoton images of mouse pulmonary alveoli are processed and used to investigate their microenvironment.
2. Experimental Setup
The experimental setup is shown in Figure 1. A mode-locked Ti:Sapphire laser (Tsunami, Spectra-Physics, Mountain View, CA, USA) was used as the excitation source for the real-time multidepth multimodal multiphoton microscopy. It generates pulses with 80 MHz repetition rate and 100 fs duration, and the center wavelength is tunable between 700 and 1000 nm. A Faraday isolator was applied to minimize back-reflection. The power and the polarization of the laser source could be tuned by rotating the half-wave plates (WPH05M-850, Thorlabs, Newton, NJ, USA) placed before (HWP1) and after (HWP2) the Faraday isolator, respectively. A polarizing beam splitter (PBS1, PBS102, Thorlabs) split the fundamental beam into two arms (ARM1, ARM2). ARM1 was p-polarized (shown in red), and ARM2 was s-polarized (shown in blue). Objectives O1 and O2 (40 × 0.75 NA, dry, UPLFLN, Olympus, Hong Kong, China) were paired to adjust the divergence of ARM2, by fine-tuning their relative position along the optical axis with a stage. Lenses L1 and L2 (AD406-B, AD410-B, LBTEK, fL1 = 50 mm, fL2 = 150 mm) expanded the beam of ARM2 to fit the back-aperture of O1. PBS2 (PBS102, Thorlabs) combined ARM1 and ARM2. Spatial synchronizations of two arms were achieved by using similar methods to those described in [18]. We placed a quarter-wave plate (QWP, WPQ05M-850, Thorlabs) and an EOM (EO-AM-NR-C1, Thorlabs) after PBS2. A function generator (SDG1032X, Siglent) was used to modulate the EOM. When the function generator applied a 4/λ voltage, the EOM and QWP worked as a half-wave plate, rotating the polarization of incoming beams by 90 degrees. When applying −4/λ voltage, the polarization of incoming beams stays unchanged. PBS3 (PBS102, Thorlabs) was placed after the EOM to remove p-polarized components. Switching the voltages applied to EOM between ±4/λ rapidly (frequency is 250 kHz) will obtain two s-polarized spatially overlapped and temporally interlaced beams. The rise time of the function generator was <4 ns, and thus crosstalk between ARM1 and ARM2 was avoidable. All components mentioned above constitute the EOM-based fast-switching light paths.
The beams were scanned by a pair of galvanometer-driven dual-axis scanning mirrors (GVS002, Thorlabs, Newton, NJ, USA) at the focal plane (i.e., scan plane) of a scan lens (SL, AC254-045-B, Thorlabs, fSL = 45 mm). Lenses L3 and L4 (AD410-B, AD406-B, LBTEK, fL3 = 150 mm, fL4 = 50 mm), along with lenses L5 and L6 (AD406-B, AD408-B, LBTEK, fL5 = 50 mm, fL6 = 100 mm), constitute two 4f optical systems, imaging the pupil of objective O2 to the middle plane of the scanning mirrors. After being focused by the scan lens, the beams were sent into a modified inverted microscope (IX71, Olympus, Tokyo, Japan), with some of its optical elements replaced to match the NIR wavelength of the excitation source. In the microscope, the beams were collected and collimated by a tube lens (TL, Olympus, Tokyo, Japan, fTL = 180 mm), which constituted another 4f optical system with the scan lens, imaging the middle plane of the scanning mirrors to the pupil of a 20 × 1.00 NA water immersion objective (O3, XLUMPLFLN, Olympus, Tokyo, Japan). The three 4f optical systems ensured that the beams of both ARM1 and ARM2 fit the back-aperture of O3 with the same sizes, and the pupil of O2, scanner, and pupil of O3 were mutually conjugate. The beams were then collected and focused into the samples by O3. The focal planes in the samples had different depths for ARM1 and ARM2, because of their different divergences. As shown in the inset of Figure 1, when ARM2 is convergent (blue) and divergent (green), its focal plane will be shallower and deeper than that of ARM1, respectively. Samples were placed on a three-dimensional motorized stage. The signal from samples was collected by the objective O3 in the epi-direction, separated from the beams by a long-pass dichroic mirror (DM1, FF705-Di01, Semrock, Lake Forest, IL, USA) and detected by two channels (CH1, CH2) equipped with photomultiplier tubes (PMT1, PMT2, R6357, Hamamatsu, Hamamatsu-shi, Japan). Another long-pass dichroic mirror (DM2, FF470-Di01, Semrock, Lake Forest, IL, USA) and two bandpass filters (F1, F2, FF02-435/40, FF01-565/133, Semrock, Lake Forest, IL, USA) were applied to separate signals at different emission regions and minimize crosstalk between different channels. The achieved wavelength ranges were 415–455 nm and 495–635 nm for CH1 and CH2, respectively. Labview-based home-made microscope software simultaneously controlled the scanner and restored mesh-like images acquired from different detection channels and beams with different divergences. Electrical devices used in the setup are commercially available, and wiring can be easily operated by researchers with limited knowledge of electrical engineering.
We demonstrate the detailed operating principle of the imaging system in detail in Figure 2. As shown in Figure 2a, the pixel clock of 2 μs period from the microscope works as an external trigger for the function generator. The function generator works in N-cycle burst mode. As shown in Figure 2b, the period of generated pulse train is 4 μs with a duty cycle of 50%. Since the EOM is modulated by the pulse train, fine-tuning the temporal position of the pulse train will ensure that only one arm in the fast-switching light paths is turned on in a pixel dwell time (2 μs). Thus, different columns of a multiphoton image correspond to focal planes of different depths, which is called pixel-to-pixel focus-switching.
Figure 2c shows real-time multidepth two-photon fluorescence image of proflavine in aqueous solution. The wavelength of the mode-locked Ti:Sapphire laser was tuned to 870 nm. The detection channel CH2 was turned on to collect autofluorescence in the range of 495–635 nm. ARM1 was focused to the surface of the proflavine solution, and ARM2 was focused to the depth of 5 μm beneath the surface. The depth offset of ARM2 was preset with the help of the Z value of the sample stage. The average powers of both ARM1 and ARM2 at the focal planes were ~5 mW, but the average power of ARM2 was slightly higher, to compensate for signal intensity loss caused by extra dispersion from dispersive elements (i.e., L1, L2, L3, L4, O1, O2). Adjacent two pixels in the same line have different intensities, showing that they were acquired at different focal planes. Figure 2d,e are autofluorescence images extracted from Figure 2c, corresponding to the depths of 0 and 5 μm, respectively. They show that the imaging system is effective in performing real-time multidepth multiphoton microscopy. Note that only one detector is needed for each imaging modality, simplifying implementation complexity. Full width at half-maximum (FWHM) axial resolutions of both ARM1 and ARM2 were approximately 1.5 μm. Measurements were taken by using fluorescent beads, when only ARM1 or ARM2 was turned on. The results show that axial resolution can be maintained when changing the divergence of beam collected by objective O3 in a small range.
3. Results and Discussion
The capability of the real-time multidepth multiphoton microscopy was demonstrated by using freshly excised mouse lung tissues. As shown in Figure 3, the lower respiratory tract, along the proximal-distal axis, includes extrapulmonary airways (i.e., trachea and primary bronchi), intrapulmonary airways (i.e., lobar bronchi, segmental bronchi, subsegmental bronchi, and bronchioles), and alveolar regions.
At the distal end of the lower respiratory tract lie the alveolar regions. We first describe the structure of mouse alveolar regions and available multiphoton imaging modalities. Alveolar regions are composed of alveoli and a network of capillaries. Alveoli are small air sacs inside lungs, allowing air exchange. Along the luminal surface, an alveolus is lined by a cell layer of squamous type I pneumocytes and cuboidal type II pneumocytes. Type I pneumocytes provide an extensive surface area for air exchange, and type II pneumocytes secrete pulmonary surfactant proteins. The pneumocytes are anchored to the subjacent epithelial basement membrane, which is apposed and often fused to the endothelial basement membrane of a surrounding capillary. Endothelial cells of the capillary adhere closely to the endothelial basement membrane. There is interstitium between the two basement membranes, where a few stromal cells (e.g., fibroblasts) may be present [28]. The epithelial basement membrane, interstitium, and endothelial basement membrane compose the extracellular matrix of an alveolus. Two main components of the extracellular matrix are collagen and elastin fibers, which are responsible for the tensile strength and elastic modulus of the alveoli, respectively. Collagen and elastin fibers can be revealed by contrast from SHG and autofluorescence, respectively. Fibroblasts in the interstitium keep synthesizing ground substance, as well as secreting collagen and elastin fibers, to maintain the integrity of the extracellular matrix. Pulmonary diseases, e.g., hyperplasia, fibrosis, and neoplasm, will cause prominent structural damage to the alveolar extracellular matrix. Besides the main structures, a few macrophages also reside in the alveoli, which can be imaged by autofluorescence predominantly from lipids and flavins [29]. Accumulation and migration of macrophages may indicate inflammation or asthma. In conclusion, researchers can use multiphoton imaging to study the morphologies of mouse alveoli for identifying structure-associated diseases and to observe cellular dynamics for monitoring physiological processes. Furthermore, because of their similar structures, mouse alveoli are good models for further investigation of human alveoli [28,30].
Simultaneously captured multidepth multiphoton images of mouse pulmonary alveoli are shown in Figure 4. The mouse was sacrificed by using cervical dislocation. Its lung lobes were removed immediately and kept in chilled phosphate-buffered saline until imaging. Animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Hebei University of Technology.
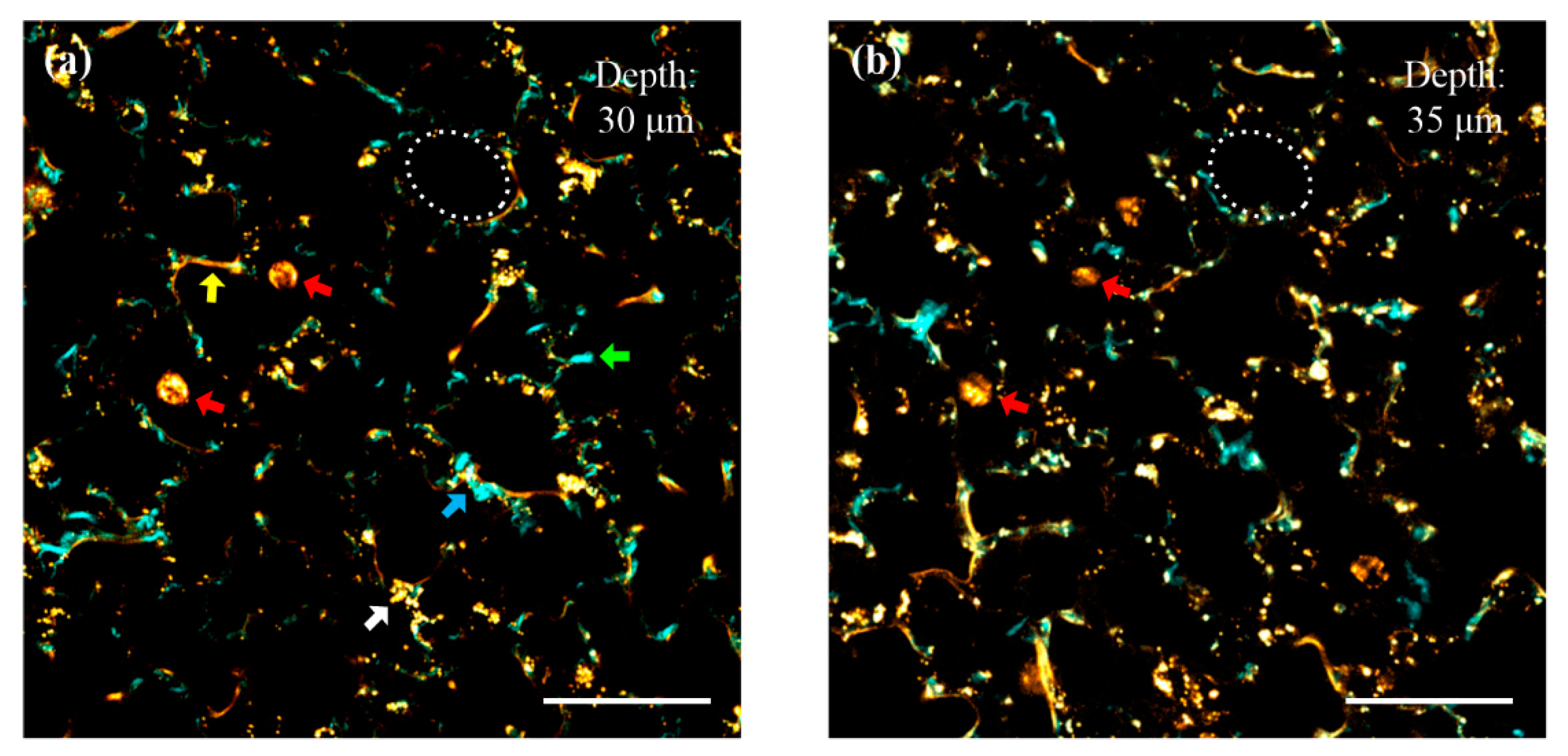
The wavelength of the mode-locked Ti:Sapphire laser was kept at 870 nm. The detection channel CH1 was turned on to collect SHG around 435 nm, and CH2 was applied to detect autofluorescence in the range of 495–635 nm. At the focal planes, both ARM1 and ARM2 have average powers of approximately 10 mW. The images were acquired at the depths of 30 μm (ARM1) and 35 μm (ARM2) into the pulmonary parenchyma. Cyan and yellow pixels in the images were captured from CH1 and CH2, respectively. As shown in Figure 4a, lumens of alveoli filled with air are apparent, and the cyan pixels may be predominantly contributed by prominent SHG from collagen fibers in the alveolar extracellular matrix, according to their typical bundle-like structures. The green arrow indicates a notable bundle of collagen fibers. The yellow pixels denoted by the yellow arrow are attributed to be elastin fibers, according to their typical rope-like structures. The blue arrow depicts that collagen bundles spiral around elastin fibers to form a condensed composite structure. This structural feature confirms that elastin fibers and collagen bundles are actually separate structures, though their signals seem to overlap in some pixels. The round-shaped yellow objects indicated by the red arrows may correspond to macrophages. The size of a macrophage is approximately 10–15 μm. The endogenous contrasts of elastin fibers and macrophages are predominantly contributed by autofluorescence emission at 495 to 635 nm, which is consistent with previously published data [31]. Gathered granules denoted by the white arrow are most likely derived from autofluorescence of surfactant proteins along luminal surfaces (probably inside the cytoplasm of type II pneumocytes). Scattered granules around collagen and elastin fibers are probably contributed by autofluorescence derived from lipopigments and flavins in fibroblasts [32]. The experimental results confirm that the imaging system is capable of revealing the microenvironment of mouse pulmonary alveoli. Figure 4b is an image obtained at the depth of 35 μm, which has the same biological components as Figure 4a. The white circles in Figure 4a,b outline the same alveolus, indicating that the focal planes switch in the axial direction without tilt. Note that images obtained at different depths have similar image qualities, proving that spherical aberrations introduced by objectives O1 and O2 are negligible. The red arrows in Figure 4b denote the same two macrophages indicated in Figure 4a, but they are imaged at different sections. This shows that the imaging system is capable of localizing biological objects in three dimensions, and it has the potential for monitoring in vivo cellular dynamics in the axial direction.
4. Conclusions
Real-time multidepth label-free TPF and SHG microscopy is achieved by using pixel-to-pixel focus-switching. It provides a cost-effective and convenient solution to modify a commercial single-focal-plane multiphoton microscope for the real-time multidepth imaging of scattering specimens. At 870 nm excitation, mouse pulmonary alveoli images acquired at different focal planes distinctly reveal the microenvironment by TPF from elastin fibers and SHG from collagen fibers, providing a tool for the investigation of structure-associated diseases. Visualization of macrophages residing inside apparent alveolar lumens by autofluorescence shows that the imaging system can localize biological objects in three dimensions. Although the offset of ARM2 was set to 5 μm in demonstrations, a range of at least ±50 μm can be achieved for different applications. Note that the pixel dwell time of the system is limited to 2 μs, only due to the restricted scanning speed of galvanometer-scanner. Applying a resonant-scanner (e.g., LSK-GR12, Thorlabs) will greatly improve the scanning speed in lateral directions, enabling three-dimensional monitoring of in vivo physiological processes. Meanwhile, objectives O1 and O2 can be replaced by a deformable mirror for fine-tuning beam divergence in future research.
Author Contributions
Conceptualization, Y.Q. and Y.X.; methodology, Y.Q., D.C., and Y.X.; software, Y.Q.; validation, Y.Q.; formal analysis, Y.Q. and Y.X.; investigation, Y.Q., D.C.; resources, Y.Q.; data curation, Y.Q.; writing—original draft preparation, Y.Q.; writing—review and editing, D.C. and Y.X.; visualization, Y.Q.; supervision, D.C., Y.X.; project administration, Y.X.; funding acquisition, Y.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China, grant numbers 61675057, 61975050, 61475040.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Richter, V.; Piper, M.; Wagner, M.; Schneckenburger, H. Increasing resolution in live cell microscopy by structured illumination (SIM). Appl. Sci. 2019, 9, 1188. [Google Scholar] [CrossRef] [Green Version]
- Ustione, A.; Piston, D.W. A simple introduction to multiphoton microscopy. J. Microsc. 2011, 243, 221–226. [Google Scholar] [CrossRef]
- Xia, F.; Wu, C.; Sinefeld, D.; Li, B.; Qin, Y.; Xu, C. In vivo label-free confocal imaging of the deep mouse brain with long-wavelength illumination. Biomed. Opt. Express 2018, 9, 6545–6555. [Google Scholar] [CrossRef]
- Sato, R.; Shimizu, Y.; Chen, C.; Matsukuma, H.; Gao, W. Investigation and improvement of thermal stability of a chromatic confocal probe with a mode-locked femtosecond laser source. Appl. Sci. 2019, 9, 4084. [Google Scholar] [CrossRef] [Green Version]
- Richter, V.; Bruns, S.; Bruns, T.; Weber, P.; Wagner, M.; Cremer, C.M.; Schneckenburger, H. Axial tomography in live cell laser microscopy. J. Biomed. Opt. 2017, 22, 091505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Shimizu, Y.; Saito, T.; Matsukuma, H.; Gao, W. Measurement uncertainty analysis of a stitching linear-scan method for the evaluation of roundness of small cylinders. Appl. Sci. 2020, 10, 4750. [Google Scholar] [CrossRef]
- Chen, C.; Sato, R.; Shimizu, Y.; Nakamura, T.; Matsukuma, H.; Gao, W. A method for expansion of Z-directional measurement range in a mode-locked femtosecond laser chromatic confocal probe. Appl. Sci. 2019, 9, 454. [Google Scholar] [CrossRef] [Green Version]
- Mandras, N.; Alovisi, M.; Roana, J.; Crosasso, P.; Luganini, A.; Pasqualini, D.; Genta, E.; Arpicco, S.; Banche, G.; Cuffini, A.; et al. Evaluation of the bactericidal activity of a hyaluronic acid-vehicled clarithromycin antibiotic mixture by confocal laser scanning microscopy. Appl. Sci. 2020, 10, 761. [Google Scholar] [CrossRef]
- Xia, F.; Wu, C.; Sinefeld, D.; Li, B.; Qin, Y.; Xu, C. In vivo label-free confocal imaging of adult mouse brain up to 1.3-mm depth with NIR-II illumination. In Proceedings of the SPIE Photonics West BiOS 2019, San Francisco, CA, USA, 2–7 February 2019. [Google Scholar]
- Zipfel, W.R.; Williams, R.M.; Webb, W.W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003, 21, 1369–1377. [Google Scholar] [CrossRef]
- Carriles, R.; Schafer, D.N.; Sheetz, K.E.; Field, J.J.; Cisek, R.; Barzda, V.; Sylvester, A.W.; Squier, J.A. Invited Review Article: Imaging techniques for harmonic and multiphoton absorption fluorescence microscopy. Rev. Sci. Instrum. 2009, 80, 081101. [Google Scholar] [CrossRef] [Green Version]
- Alovisi, M.; Pasqualini, D.; Carpegna, G.; Comba, A.; Moccia, E.; Multari, S.; Dioguardi, M.; Scotti, N.; Berutti, E. The influence of brushing movement on geometrical shaping outcomes: A micro‐CT study. Appl. Sci. 2020, 10, 4805. [Google Scholar] [CrossRef]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Li, B.; Xia, F.; Xia, Y.; Xu, C. Time-lens based multi-color background-free coherent anti-Stokes Raman scattering microscopy. In Proceedings of the SPIE Photonics West BiOS 2019, San Francisco, CA, USA, 2–7 February 2019. [Google Scholar]
- Ouzounov, D.G.; Wang, T.; Wang, M.; Feng, D.D.; Horton, N.G.; Cruz-Hernández, J.C.; Cheng, Y.-T.; Reimer, J.; Tolias, A.S.; Nishimura, N.; et al. In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nat. Methods 2017, 14, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wu, C.; Wang, M.; Charan, K.; Xu, C. An adaptive excitation source for high-speed multiphoton microscopy. Nat. Methods 2020, 17, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Rohrbacher, A.; Olarte, O.E.; Villamaina, V.; Loza-Alvarez, P.; Resan, B. Multiphoton imaging with blue-diode-pumped SESAM-modelocked Ti:sapphire oscillator generating 5 nJ 82 fs pulses. Opt. Express 2017, 25, 10677–10684. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, B.; Xia, F.; Xia, Y.; Xu, C. Multi-color background-free coherent anti-Stokes Raman scattering microscopy using a time-lens source. Opt. Express 2018, 26, 34474–34483. [Google Scholar] [CrossRef] [PubMed]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by phasor fluorescence lifetime microscopy of NADH. Sci. Rep. 2012, 2, 568. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Rico-Jimenez, J.J.; Zhang, C.; Alex, A.; Chaney, E.J.; Barkalifa, R.; Spillman, D.R., Jr.; Marjanovic, M.; Arp, Z.; Hood, S.R.; et al. Simultaneous label-free autofluorescence and multi-harmonic imaging reveals in vivo structural and metabolic changes in murine skin. Biomed. Opt. Express 2019, 10, 5431–5444. [Google Scholar] [CrossRef]
- You, S.; Tu, H.; Chaney, E.J.; Sun, Y.; Zhao, Y.; Bower, A.J.; Liu, Y.-Z.; Marjanovic, M.; Sinha, S.; Pu, Y.; et al. Intravital imaging by simultaneous label-free autofluorescence-multiharmonic microscopy. Nat. Commun. 2018, 9, 2125. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Handley, M.; Carles, G.; Harveya, A.R. Advances in 3D single particle localization microscopy. APL Photon. 2019, 4, 060901. [Google Scholar] [CrossRef] [Green Version]
- Velmurugan, R.; Chao, J.; Ram, S.; Ward, E.S.; Ober, R.J. Intensity-based axial localization approaches for multifocal plane microscopy. Opt. Express 2017, 25, 3394–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahmasbi, A.; Ram, S.; Chao, J.; Abraham, A.V.; Tang, F.W.; Ward, E.S.; Ober, R.J. Designing the focal plane spacing for multifocal plane microscopy. Opt. Express 2014, 22, 16706–16721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajj, B.; Oudjedi, L.; Fiche, J.-B.; Dahan, M.; Nollmann, M. Highly efficient multicolor multifocus microscopy by optimal design of diffraction binary gratings. Sci. Rep. 2017, 7, 5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Li, Q.; Xia, Y.; Liu, B.; Zhang, S. Construction and application of femtosecond laser two-photon fluorescence microscopy system. J. Harbin Inst. Technol. 2015, 47, 1–5. [Google Scholar]
- Wang, S.; Qin, Y.; Guo, M.; Zhang, S.; Xia, Y. High speed 3D two-photon fluorescence microscopy by femtosecond laser pulses. In Proceedings of the LIDAR Imaging Detection and Target Recognition 2017, Changchun, China, 23–25 July 2017. [Google Scholar]
- Barkauskas, C.E.; Chung, M.-I.; Fioret, B.; Gao, X.; Katsura, H.; Hogan, B.L.M. Lung organoids: Current uses and future promise. Development 2017, 144, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Abraham, T.; Hirota, J.A.; Wadsworth, S.; Knight, D.A. Minimally invasive multiphoton and harmonic generation imaging of extracellular matrix structures in lung airway and related diseases. Pulm. Pharmacol. Ther. 2011, 24, 487–496. [Google Scholar] [CrossRef]
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, W.R.; Williams, R.M.; Christie, R.; Nikitin, A.Y.; Hyman, B.T.; Webb, W.W. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc. Natl. Acad. Sci. USA 2003, 100, 7075–7080. [Google Scholar] [CrossRef] [Green Version]
- Filippi, A.; Sasso, E.D.; Iop, L.; Armani, A.; Gintoli, M.; Sandri, M.; Gerosa, G.; Romanato, F.; Borile, G. Multimodal label-free ex vivo imaging using a dual-wavelength microscope with axial chromatic aberration compensation. J. Biomed. Opt. 2018, 23, 091403. [Google Scholar]
Figure 1.
Experimental setup of a real-time multidepth multiphoton imaging system. The excitation beams are shown in red and blue, and generated signals at different wavelength ranges are shown in green and yellow. The electrical paths are indicated in dashed black lines. HWP1, HWP2, half-wave plates; FI, Faraday isolator; PBS1, PBS2, PBS3, polarizing beam splitters; M, mirror; L1, L2, L3, L4, L5, L6, lenses; O1, O2, O3, objectives; QWP, quarter-wave plate; EOM, electro-optic modulator; B, blocker; SL, scan lens; TL, tube lens; DM1, DM2, long pass dichroic mirrors; F1, F2, bandpass filters; PMT1, PMT2, photomultiplier tubes. This figure is not to scale.
Figure 1.
Experimental setup of a real-time multidepth multiphoton imaging system. The excitation beams are shown in red and blue, and generated signals at different wavelength ranges are shown in green and yellow. The electrical paths are indicated in dashed black lines. HWP1, HWP2, half-wave plates; FI, Faraday isolator; PBS1, PBS2, PBS3, polarizing beam splitters; M, mirror; L1, L2, L3, L4, L5, L6, lenses; O1, O2, O3, objectives; QWP, quarter-wave plate; EOM, electro-optic modulator; B, blocker; SL, scan lens; TL, tube lens; DM1, DM2, long pass dichroic mirrors; F1, F2, bandpass filters; PMT1, PMT2, photomultiplier tubes. This figure is not to scale.
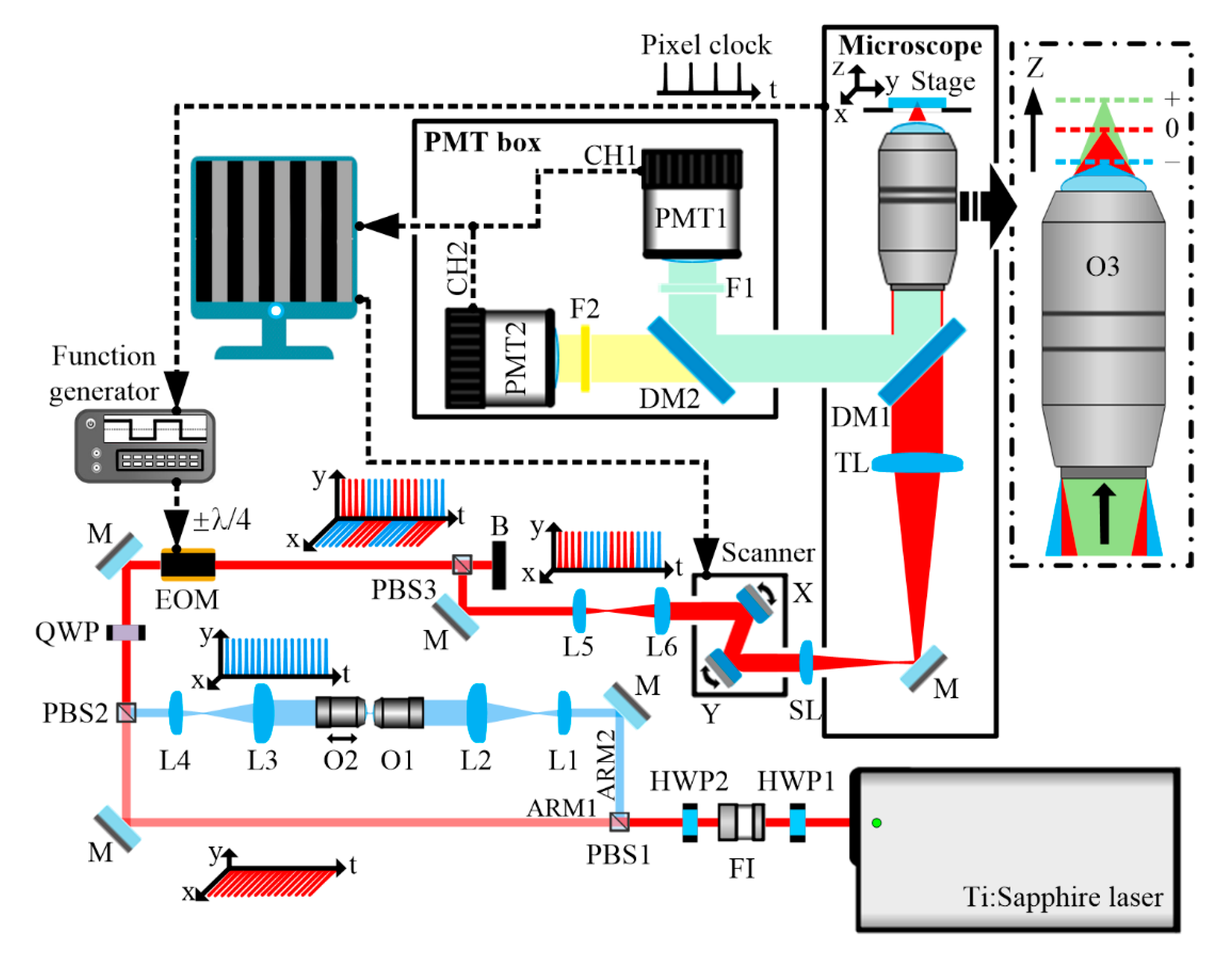
Figure 2.
Demonstration of real-time multidepth multiphoton imaging using pixel-to-pixel focus-switching, 2 μs/pixel, 200 × 200 pixels for (c), 100 × 100 pixels for panels (d,e). (a) The pixel clock from the microscope. (b) Pulse train from the function generator. (c) Autofluorescence image of proflavine in aqueous solution. Signals obtained from the depths of 0 and 5 μm are displayed in different columns of the same image. The inset is a zoomed-in view of 2 × 4 pixels. (d,e) are autofluorescence images extracted from different columns of (c), corresponding to the depths of 0 and 5 μm, respectively. The scale bars are 10 μm.
Figure 2.
Demonstration of real-time multidepth multiphoton imaging using pixel-to-pixel focus-switching, 2 μs/pixel, 200 × 200 pixels for (c), 100 × 100 pixels for panels (d,e). (a) The pixel clock from the microscope. (b) Pulse train from the function generator. (c) Autofluorescence image of proflavine in aqueous solution. Signals obtained from the depths of 0 and 5 μm are displayed in different columns of the same image. The inset is a zoomed-in view of 2 × 4 pixels. (d,e) are autofluorescence images extracted from different columns of (c), corresponding to the depths of 0 and 5 μm, respectively. The scale bars are 10 μm.
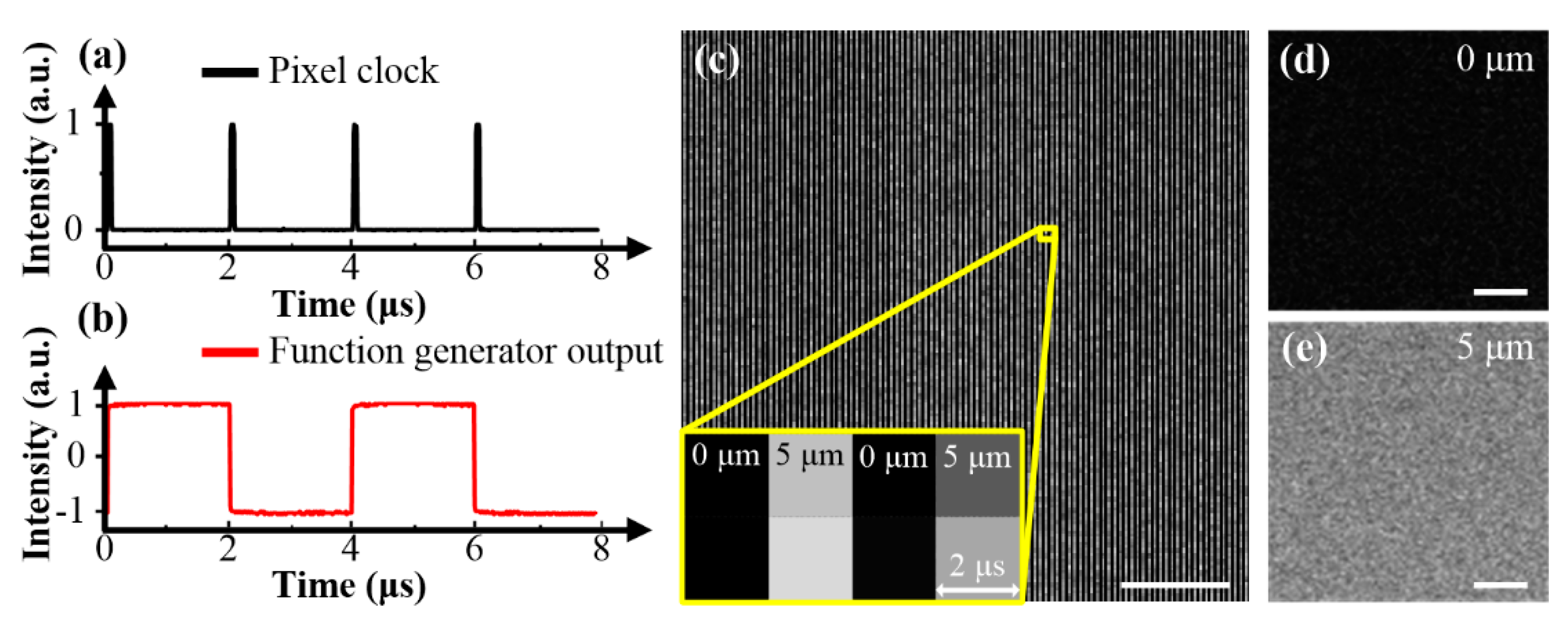
Figure 3.
Schematic of mouse lower respiratory tract. The lower respiratory tract, along the proximal-distal axis, includes extrapulmonary airways, intrapulmonary airways, and alveolar regions. The alveolar regions are composed of several distinct layers, i.e., pneumocytes, epithelial basement membrane, interstitium, endothelial basement membrane, and endothelial cells. The pneumocytes are composed of type I pneumocytes responsible for air exchange and type II pneumocytes secreting surfactant proteins. A few macrophages may be present in the alveolar lumens or interstitium. Fibroblast can be found in the interstitium.
Figure 3.
Schematic of mouse lower respiratory tract. The lower respiratory tract, along the proximal-distal axis, includes extrapulmonary airways, intrapulmonary airways, and alveolar regions. The alveolar regions are composed of several distinct layers, i.e., pneumocytes, epithelial basement membrane, interstitium, endothelial basement membrane, and endothelial cells. The pneumocytes are composed of type I pneumocytes responsible for air exchange and type II pneumocytes secreting surfactant proteins. A few macrophages may be present in the alveolar lumens or interstitium. Fibroblast can be found in the interstitium.

Figure 4.
Simultaneously acquired multiphoton images of mouse pulmonary alveoli at different depths, 2 μs/pixel, 512 × 512 pixels. (a) A pseudo-colored two-photon fluorescence (TPF) and second-harmonic generation (SHG) image acquired at the depth of 30 μm into the pulmonary parenchyma. Cyan pixels (obtained from CH1) are mainly derived from prominent SHG of collagen. The green arrow indicates a pronounced bundle of collagen fibers. Yellow pixels (obtained from CH2) are mainly derived from autofluorescence of elastin, macrophages, and surfactant proteins. The yellow arrow denotes notable elastin fibers. The white arrow indicates surfactant proteins gathered probably in the cytoplasm of a type II pneumocyte. The blue arrow shows that collagen fibers spiral around elastin fibers. (b) A pseudo-colored TPF and SHG image simultaneously acquired at the depth of 35 μm into the pulmonary parenchyma. The red arrows in (a,b) denote the sections of the same macrophages at both depths. The scale bar is 40 μm.
Figure 4.
Simultaneously acquired multiphoton images of mouse pulmonary alveoli at different depths, 2 μs/pixel, 512 × 512 pixels. (a) A pseudo-colored two-photon fluorescence (TPF) and second-harmonic generation (SHG) image acquired at the depth of 30 μm into the pulmonary parenchyma. Cyan pixels (obtained from CH1) are mainly derived from prominent SHG of collagen. The green arrow indicates a pronounced bundle of collagen fibers. Yellow pixels (obtained from CH2) are mainly derived from autofluorescence of elastin, macrophages, and surfactant proteins. The yellow arrow denotes notable elastin fibers. The white arrow indicates surfactant proteins gathered probably in the cytoplasm of a type II pneumocyte. The blue arrow shows that collagen fibers spiral around elastin fibers. (b) A pseudo-colored TPF and SHG image simultaneously acquired at the depth of 35 μm into the pulmonary parenchyma. The red arrows in (a,b) denote the sections of the same macrophages at both depths. The scale bar is 40 μm.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Qin, Y.; Chen, D.; Xia, Y. Real-Time Multidepth Multiphoton Microscopy Using Pixel-to-Pixel Focus-Switching. Appl. Sci. 2020, 10, 7173. https://doi.org/10.3390/app10207173
AMA Style
Qin Y, Chen D, Xia Y. Real-Time Multidepth Multiphoton Microscopy Using Pixel-to-Pixel Focus-Switching. Applied Sciences. 2020; 10(20):7173. https://doi.org/10.3390/app10207173
Chicago/Turabian StyleQin, Yifan, Deying Chen, and Yuanqin Xia. 2020. "Real-Time Multidepth Multiphoton Microscopy Using Pixel-to-Pixel Focus-Switching" Applied Sciences 10, no. 20: 7173. https://doi.org/10.3390/app10207173
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.