
Influence of Hard Segments on the Thermal, Phase-Separated Morphology, Mechanical, and Biological Properties of Polycarbonate Urethanes

Abstract
:1. Introduction
2. Experimental Methods
2.1. Materials
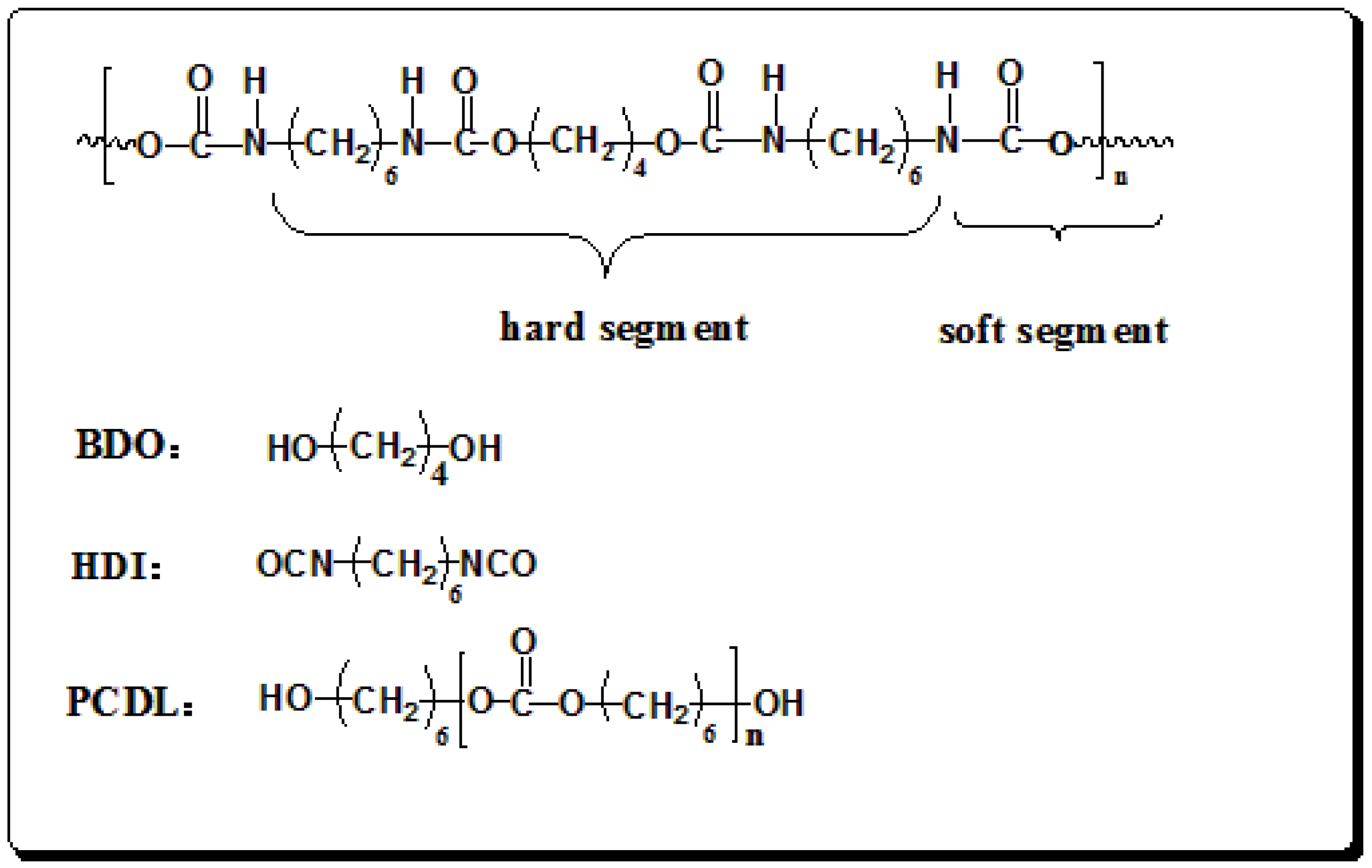
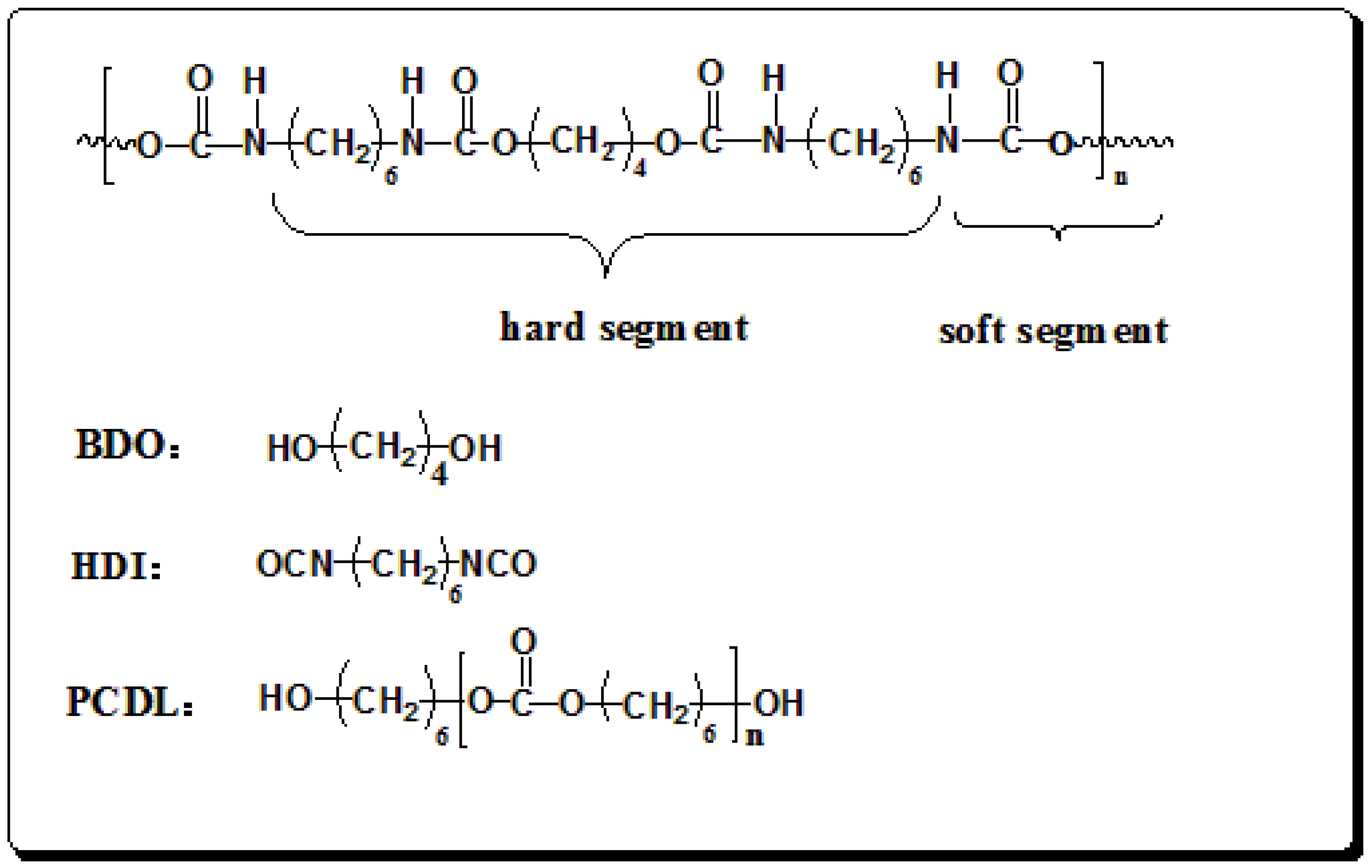
2.2. Synthesis and Preparation of Polyurethane Films
2.3. Characterization Methods
3. Results and Discussion
3.1. Molecular Weight Determination
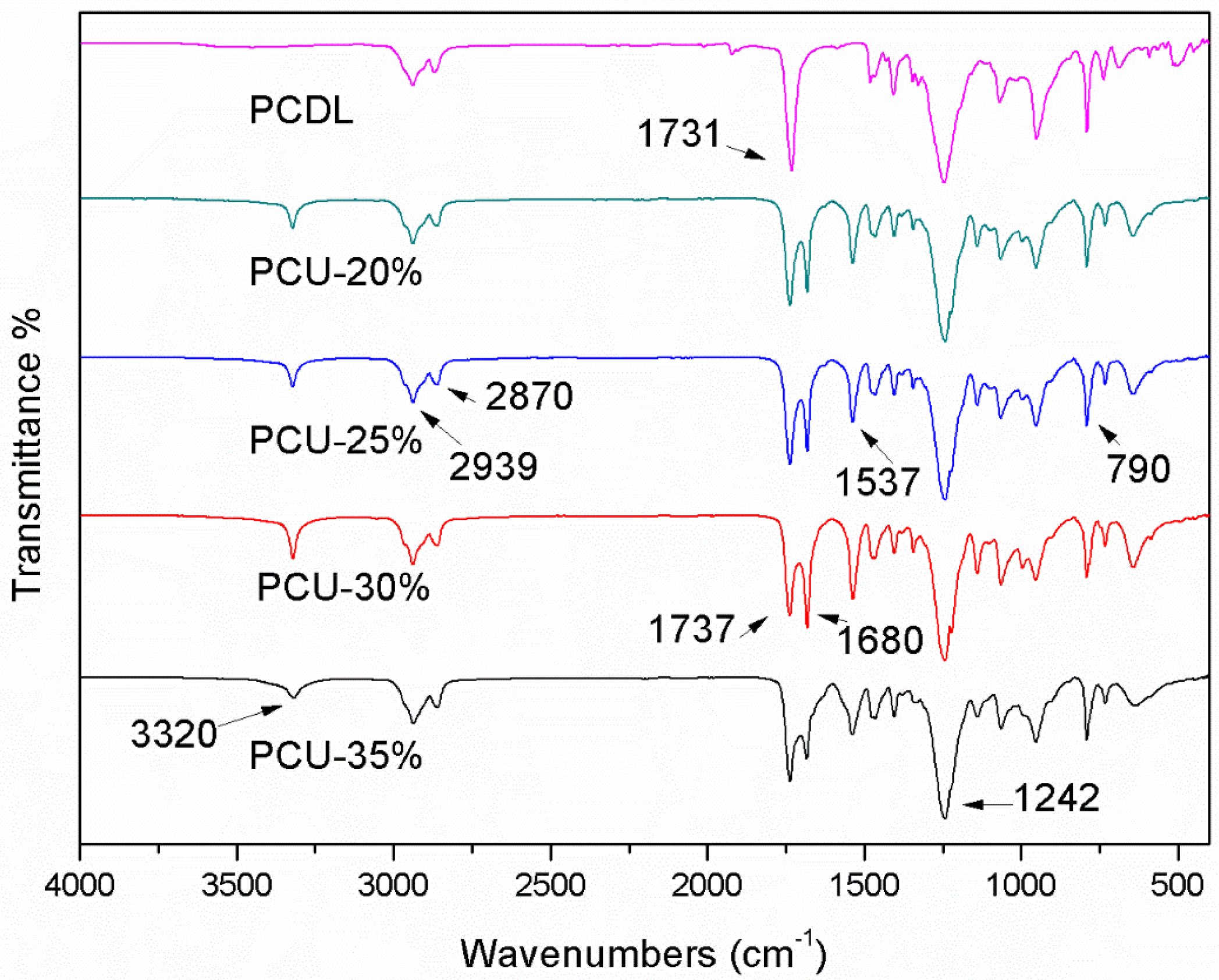
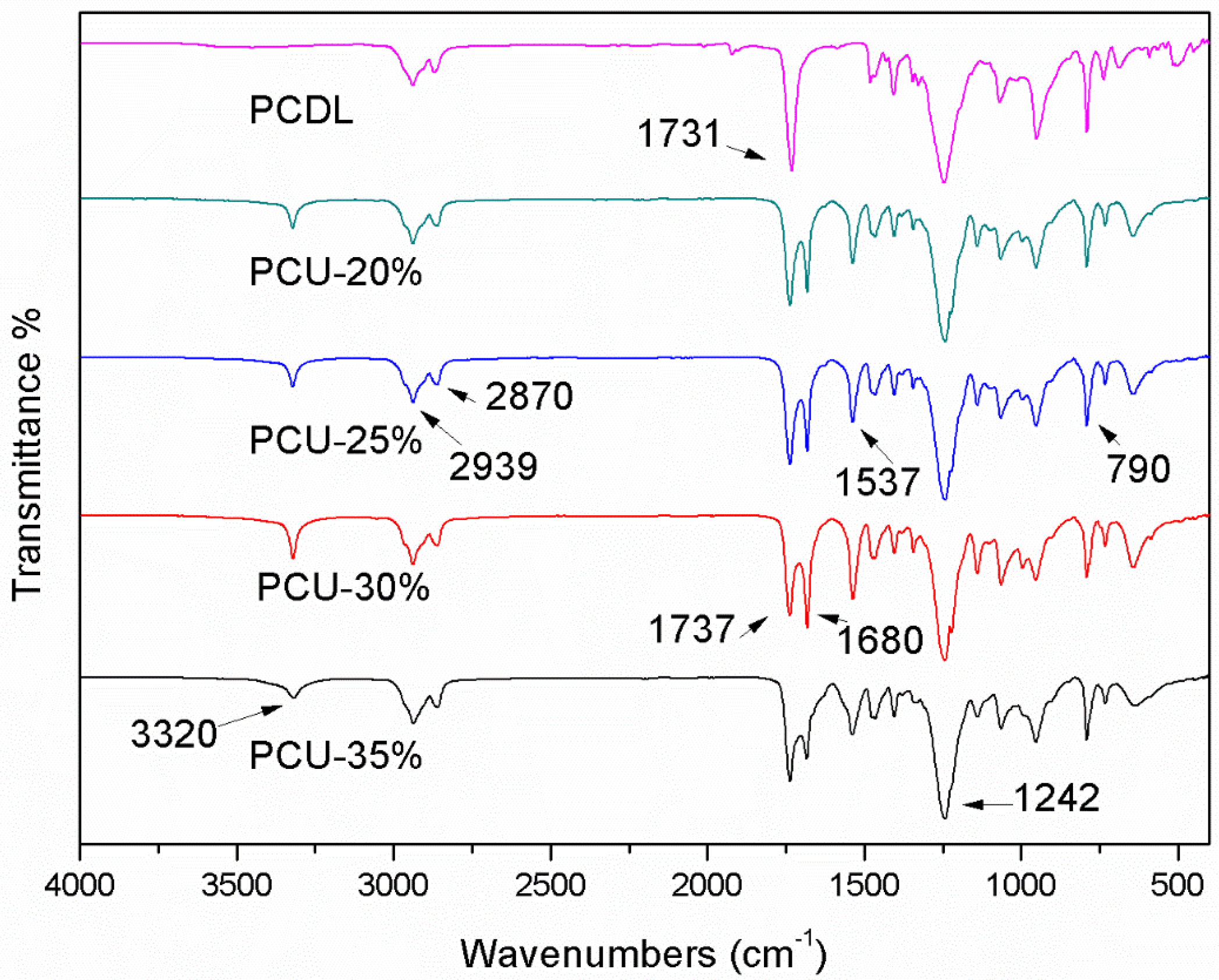
3.2. ATR-FTIR Analysis
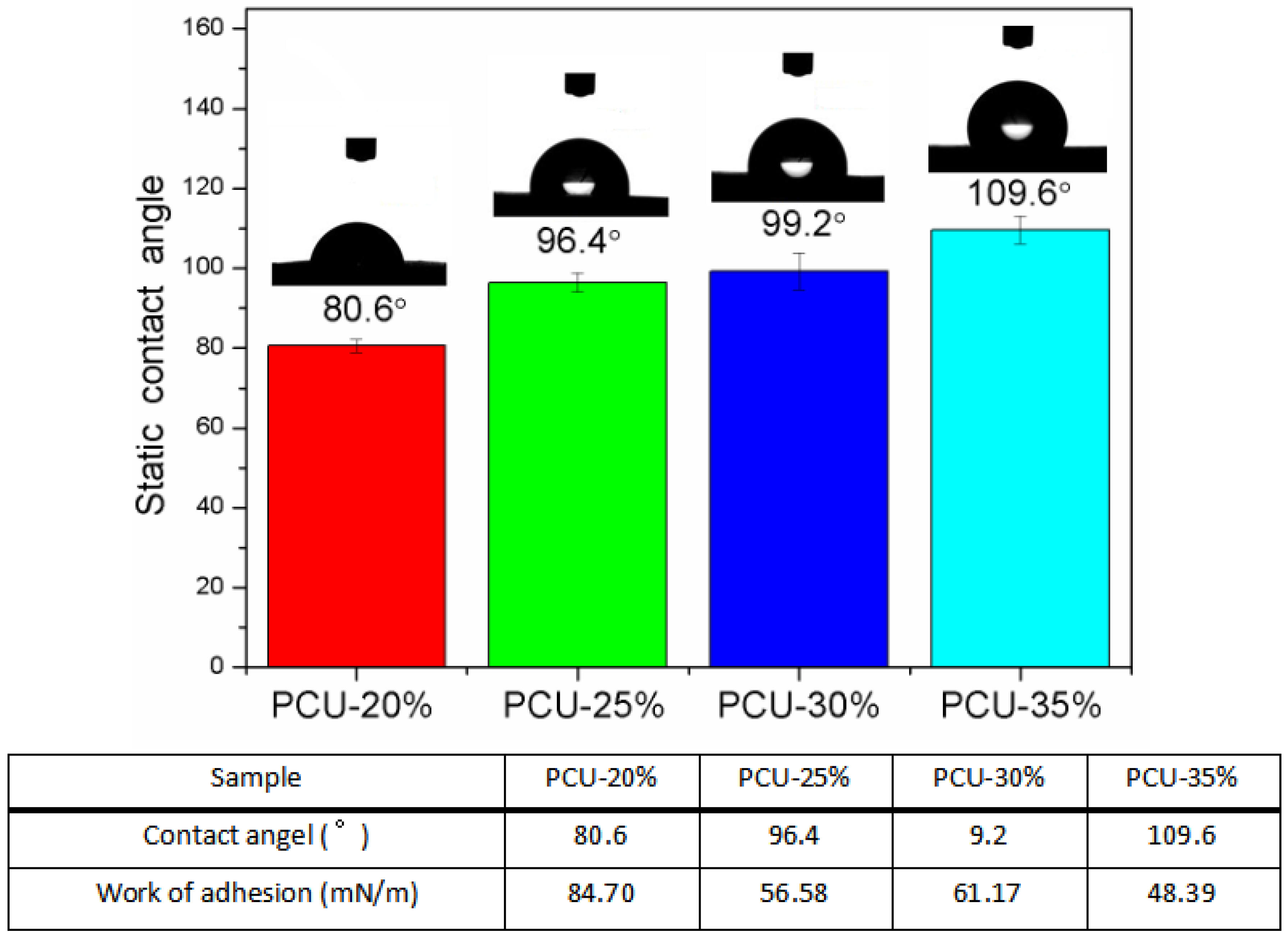
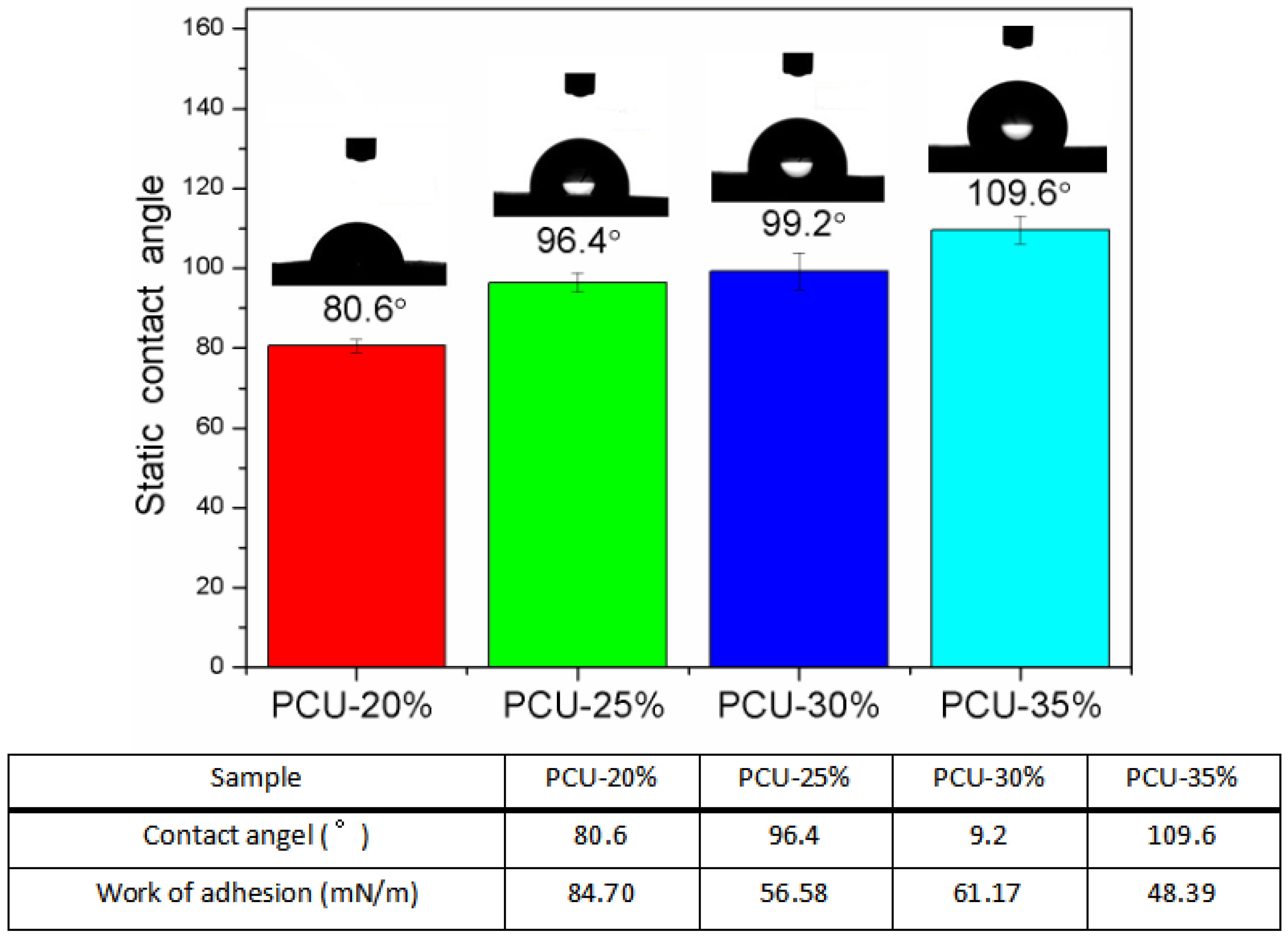
3.3. The Surface Morphology and Wettability Analysis
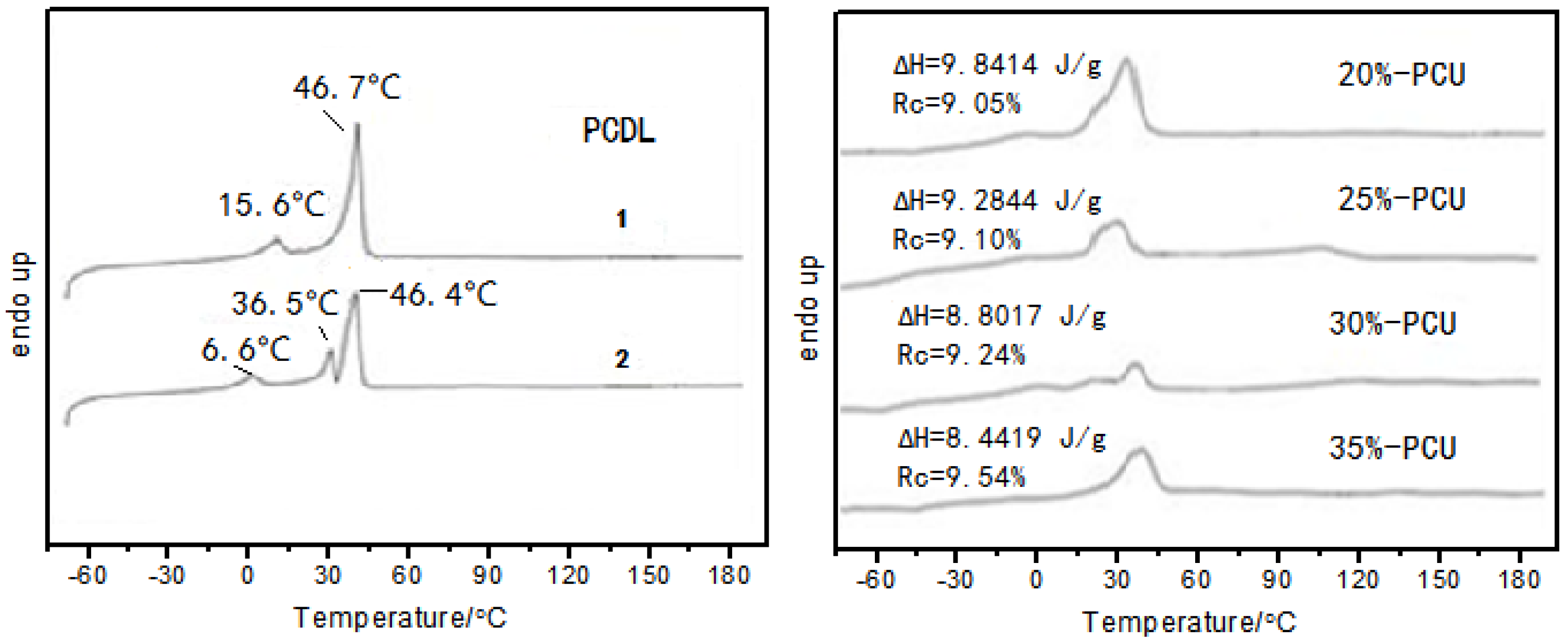
3.4. Differential Scanning Calorimetry
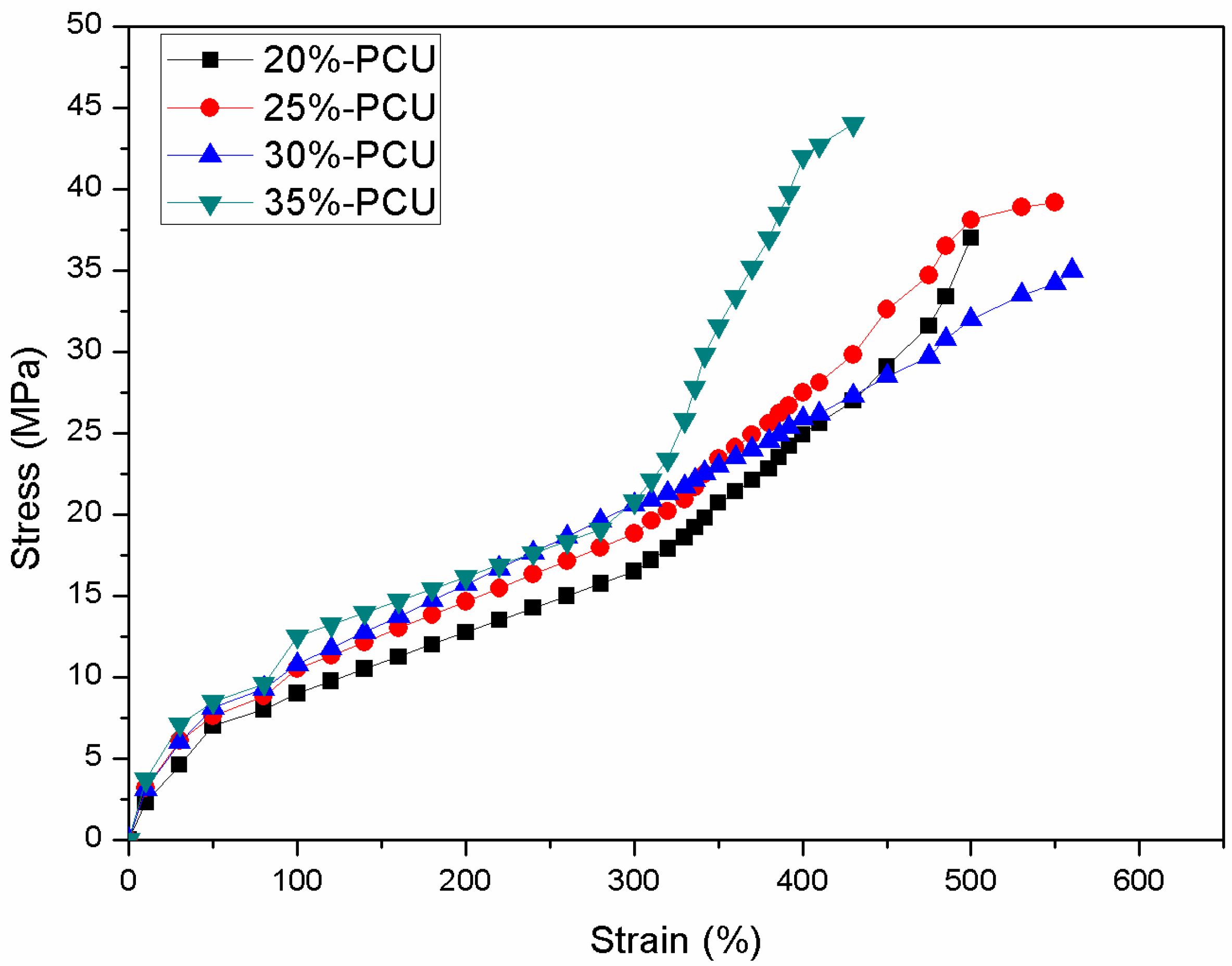
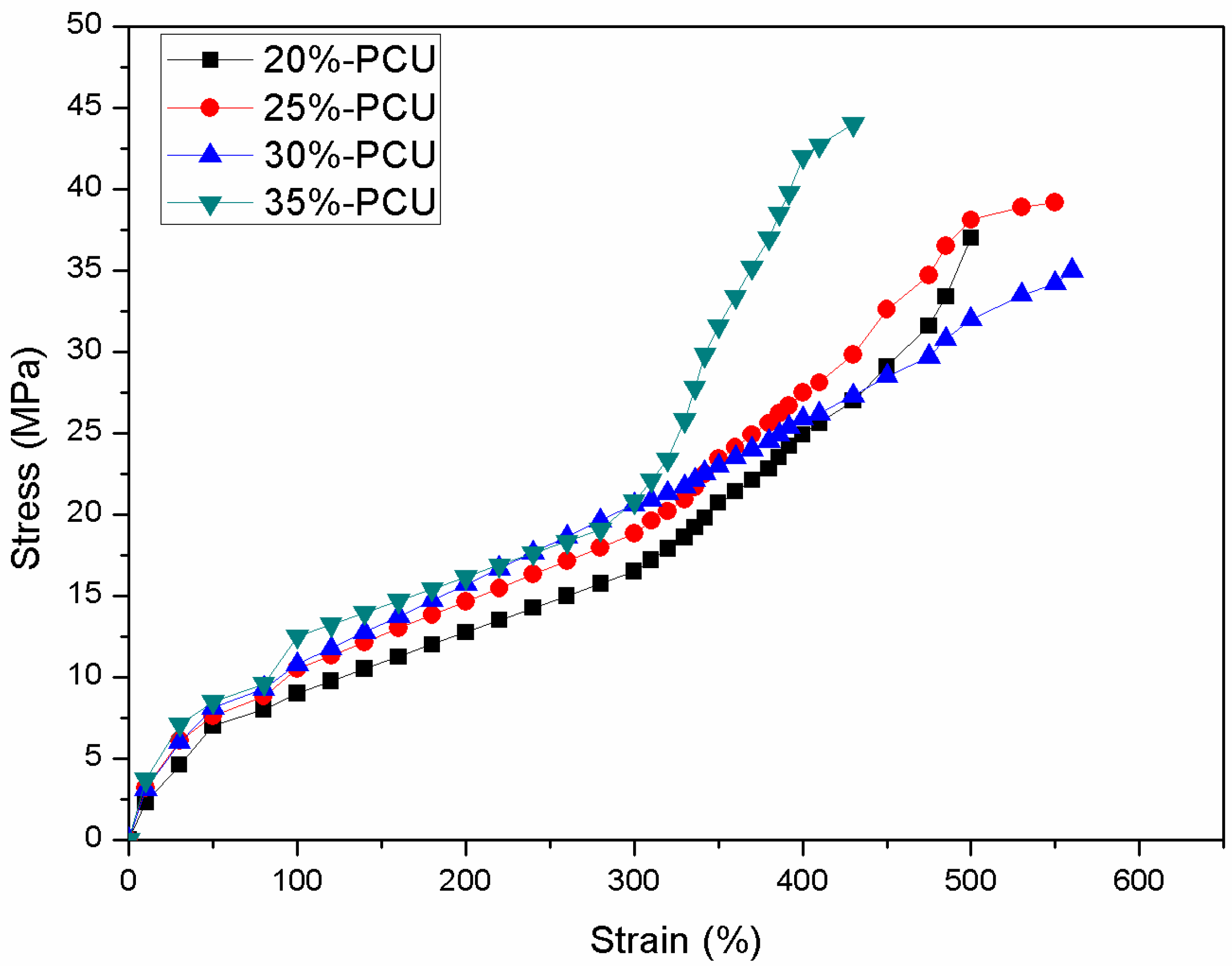
3.5. Mechanical Properties Measurement
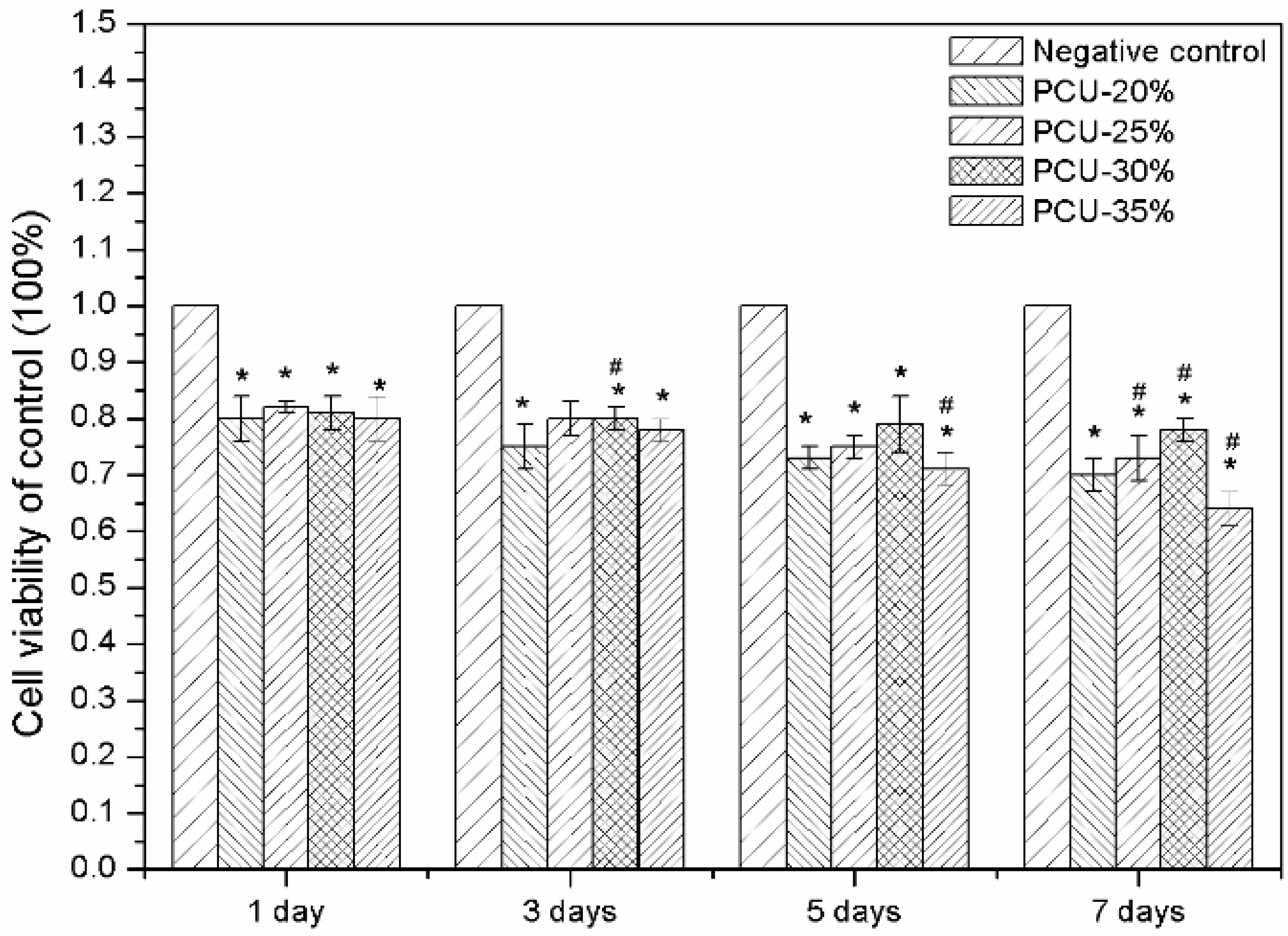
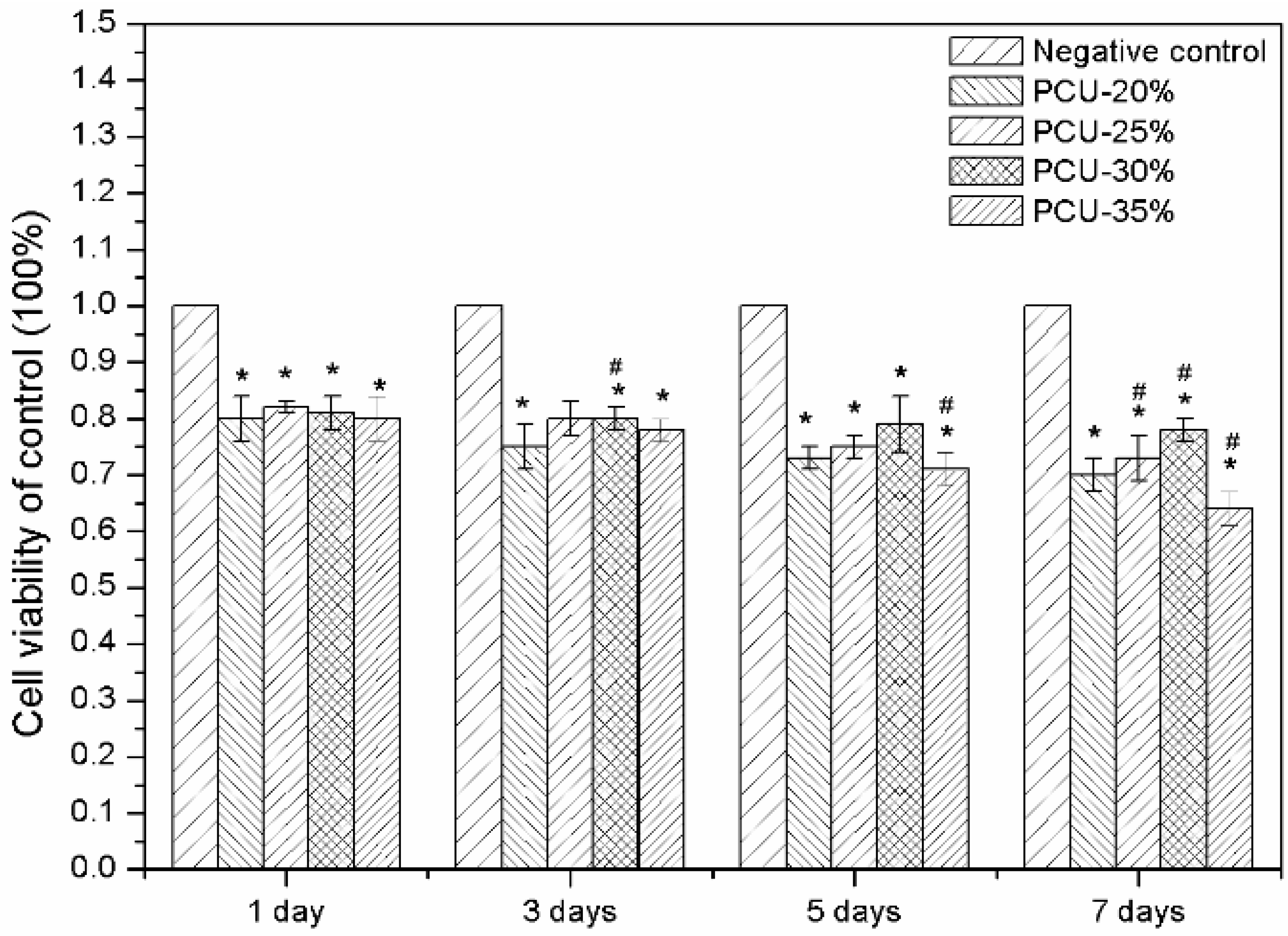
3.6. Hemolytic Experiments and Cytotoxicity Assay by MTT
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heijkants, R.G.J.C.; van Calck, R.V.; van Tienen, T.G.; de Groot, J.H. Uncatalyzed synthesis, thermal and mechanical properties of polyurethanes based on poly (ε-caprolactone) and 1,4-butane diisocyanate with uniform hard segment. J. Biomater. 2005, 26, 4219–4228. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, D.K.; Webster, D.C. Thermal stability and flame retardancy of polyurethanes. J. Prog. Polym. Sci. 2009, 34, 1068–1133. [Google Scholar] [CrossRef]
- Madhavan, K.; Reddy, B.S.R. Synthesis and characterization of polyurethane hybrids: Influence of the polydimethylsiloxane linear chain and silsesquioxane cubic structure on the thermal and mechanical properties of polyurethane hybrids. J. Appl. Polym. Sci. 2009, 113, 4052–4065. [Google Scholar] [CrossRef]
- Biemond, G.J.E.; Brasspenning, K.; Gaymans, R.J. Synthesis and selected properties of polyurethanes with monodisperse hard segments based on hexane diisocyanate and three types of chain extenders. J. Appl. Polym. Sci. 2012, 124, 1302–1315. [Google Scholar] [CrossRef]
- Guelcher, S.A.; Srinivasan, A.; Dumas, J.E.; Didier, J.E. Synthesis, mechanical properties, biocompatibility, and biodegradation of polyurethane networks from lysine polyisocyanates. J. Biomater. 2008, 29, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.D.; Farrugia, B.L.; Dargaville, T.R.; Dhara, S. Chitosan–collagen scaffolds with nano/microfibrous architecture for skin tissue engineering. J. Biomed. Mater. Res. A 2013, 101, 3482–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrag, I.C.; Woodhouse, K.A. Development of biodegradable polyurethane scaffolds using amino acid and dipeptide-based chain extenders for soft tissue engineering. J. Biomater. Sci. Polym. E 2010, 21, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, A.; Bin, X.; Purnawali, H.; Fu, Y. High performance shape memory polyurethane synthesized with high molecular weight polyol as the soft segment. J. Appl. Sci. 2012, 2, 535–548. [Google Scholar]
- Oprea, S. Structure and properties of cross-linked polyurethane copolymers. J. Adv. Polym. Technol. 2009, 28, 165–172. [Google Scholar] [CrossRef]
- Bai, H.C.; Tao, L.; Pang, Y.J.; Zhou, Y.J. Synthesis and characterization of hydroxypropyl terminated polydimethylsiloxane-polyurethane copolymers. J. Appl. Polym. Sci. 2013, 129, 2152–2160. [Google Scholar] [CrossRef]
- Li, C.; Yu, X.; Speckhard, T.A.; Cooper, S.L. Synthesis and properties of polycyanoethylmethylsiloxane polyurea urethane elastomers: A study of segmental compatibility. J. Polym. Sci. Pol. Phys. 1988, 26, 315–337. [Google Scholar] [CrossRef]
- Marcos-Fernandez, A.; Lozano, A.E.; González, L.; Rodríguez, A. Hydrogen bonding in copoly (ether-urea)s and its relationship with the physical properties. J. Macromol. 1997, 30, 3584–3592. [Google Scholar] [CrossRef]
- Špírková, M.; Pavličević, J.; Strachotaa, A.; Porebaa, R. Novel polycarbonate-based polyurethane elastomers: Composition–property relationship. J. Eur. Polym. J. 2011, 47, 959–972. [Google Scholar] [CrossRef]
- Adhikari, R.; Gunatillake, P.A.; McCarthy, S.J.; Bown, M. Low-modulus siloxane–polyurethanes. Part II. Effect of chain extender structure on properties and morphology. J. Appl. Polym. Sci. 2003, 87, 1092–1100. [Google Scholar] [CrossRef]
- Shokrolahi, F.; Yeganeh, H. Soft segment composition and its influence on phase-separated morphology of PCL/PEG-based poly (urethane urea)s. J. Iran. Polym. J. 2014, 23, 505–512. [Google Scholar] [CrossRef]
- Fragiadakis, D.; Runt, J. Molecular dynamics of segmented polyurethane copolymers: Influence of soft segment composition. J. Macromol. 2013, 46, 4184–4190. [Google Scholar] [CrossRef]
- Mingjie, H.; Wei, F.; Le, G.; Weibing, W. One-pot synthesis of hyperbranched polyols and their effects as crosslinkers on HTPB-based polyurethane. J. Polym. Bull. 2014, 71, 2671–2693. [Google Scholar] [CrossRef]
- Król, P.; Pielichowska, K.; Byczyńskia, Ł. Thermal degradation kinetics of polyurethane–siloxane anionomers. J. Thermochim. Acta 2010, 507, 91–98. [Google Scholar] [CrossRef]
- Castagna, A.M.; Fragiadakis, D.; Lee, H.; Choi, T. The role of hard segment content on the molecular dynamics of poly (tetramethylene oxide)-based polyurethane copolymers. J. Macromol. 2011, 44, 7831–7836. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, G.; Zhang, Y.; Gao, Y. Synthesis, characterization, and properties of novel polyetherester polyols and developed polyurethanes. J. Appl. Polym. Sci. 2007, 103, 417–424. [Google Scholar] [CrossRef]
- Chattopadhyay, D.K.; Raju, K. Structural engineering of polyurethane coatings for high performance applications. J. Prog. Polym. Sci. 2007, 32, 352–418. [Google Scholar] [CrossRef]
- Fu, H.; Gao, H.; Wu, G.; Wang, Y. Preparation and tunable temperature sensitivity of biodegradable polyurethane nanoassemblies from diisocyanate and poly (ethylene glycol). J. Soft Matter 2011, 7, 3546–3552. [Google Scholar] [CrossRef]
- Mondal, S.; Martin, D. Hydrolytic degradation of segmented polyurethane copolymers for biomedical applications. J. Polym. Degrad. Stab. 2012, 97, 1553–1561. [Google Scholar] [CrossRef]
- Oprea, S. Degradation of crosslinked poly (ester-urethanes) elastomers in distilled water: Influence of hard segment. J. Appl. Polym. Sci. 2012, 124, 1059–1066. [Google Scholar] [CrossRef]
- Salacinski, H.J.; Odlyha, M.; Hamilton, G.; Seifalian, A.M. Thermo-mechanical analysis of a compliant poly (carbonate-urea) urethane after exposure to hydrolytic, oxidative, peroxidative and biological solutions. J. Biomater. 2002, 23, 2231–2240. [Google Scholar] [CrossRef]
- Kultys, A.; Rogulska, M.; Pikus, S.; Skrzypiec, K. The synthesis and characterization of new thermoplastic poly (carbonate-urethane) elastomers derived from HDI and aliphatic–aromatic chain extenders. J. Eur. Polym. J. 2009, 45, 2629–2643. [Google Scholar] [CrossRef]
- Seifalian, A.M.; Salacinski, H.J.; Tiwan, A.; Edwards, A. In vivo biostability of a poly (carbonate-urea) urethane graft. J. Biomater. 2003, 24, 2549–2557. [Google Scholar] [CrossRef]
- Pergal, M.V.; Antić, V.V.; Govedarica, M.N.; Goäevac, D. Synthesis and characterization of novel urethane-siloxane copolymers with a high content of PCL-PDMS-PCL segments. J. Appl. Polym. Sci. 2011, 122, 2715–2730. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, Y.; Zhang, Z.; Ma, D. Synthesis of polycarbonate urethane elastomers and effects of the chemical structures on their thermal, mechanical and biocompatibility properties. J. Heliyon 2016, 2, e00125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Shi, J.; Zhu, R. Flexible silk fibroin films modified by genipin and glycerol. J. RSC Adv. 2015, 5, 101362–101369. [Google Scholar] [CrossRef]
- Choi, T.; Masser, K.A.; Moore, E.; Weksler, J. Segmented polyurethanes derived from novel siloxane–carbonate soft segments for biomedical applications. J. Polym. Sci. Pol. Phys. 2011, 49, 865–872. [Google Scholar] [CrossRef]
- Wang, C.B.; Cooper, S.L. Morphology and properties of segmented polyether polyurethaneureas. J. Macromol. 1983, 16, 775–786. [Google Scholar] [CrossRef]
- Oprea, S.; Potolinca, V.O.; Oprea, V. Synthesis and properties of new crosslinked polyurethane elastomers based on isosorbide. J. Eur. Polym. J. 2016, 83, 161–172. [Google Scholar] [CrossRef]
- Cassie, A.B.D.; Baxter, S. Wettability of porous surfaces. J. Trans. Faraday Soc. 1944, 40, 546–551. [Google Scholar] [CrossRef]
- Seo, J.H.; Kakinoki, S.; Inoue, Y.; Nam, K. The significance of hydrated surface molecular mobility in the control of the morphology of adhering fibroblasts. J. Biomater. 2013, 34, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Extrand, C.W.; Kumagai, Y. An experimental study of contact angle hysteresis. J. Colloid Interface Sci. 1997, 191, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Sheikhy, H.; Shahidzadeh, M.; Ramezanzadeh, B.; Noroze, F. Studying the effects of chain extenders chemical structures on the adhesion and mechanical properties of a polyurethane adhesive. J. Ind. Eng. Chem. 2013, 19, 1949–1955. [Google Scholar] [CrossRef]
- Jiang, S.; Ji, X.; An, L.; Jiang, B. Crystallization behavior of PCL in hybrid confined environment. J. Polym. 2001, 42, 3901–3907. [Google Scholar] [CrossRef]
- De Groot, J.H.; De Vrijer, R.; Wildeboer, B.S.; Spaans, C.S. New biomedical polyurethane ureas with high tear strengths. J. Polym. Bull. 1997, 38, 211–218. [Google Scholar] [CrossRef]
- Miller, J.A.; Lin, S.B.; Hwang, K.K.S.; Wu, K.S.; Gibson, P.E.; Cooper, S.L. Properties of polyether-polyurethane block copolymers: Effects of hard segment length distribution. J. Macromol. 1985, 18, 32–44. [Google Scholar] [CrossRef]
- Eceiza, A.; Martin, M.D.; De la Caba, K.; Kortaberria, G. Thermoplastic polyurethane elastomers based on polycarbonate diols with different soft segment molecular weight and chemical structure: Mechanical and thermal properties. J. Polym. Eng. Sci. 2008, 48, 297–306. [Google Scholar] [CrossRef]
- Teramoto, N.; Saitoh, Y.; Takahashi, A.; Shibata, M. Biodegradable polyurethane elastomers prepared from isocyanate-terminated poly(ethylene adipate), castor oil, and glycerol. J. Appl. Polym. Sci. 2010, 115, 3199–3204. [Google Scholar] [CrossRef]
- Chiou, B.S.; Schoen, P.E. Effects of crosslinking on thermal and mechanical properties of polyurethanes. J. Appl. Polym. Sci. 2002, 83, 212–223. [Google Scholar] [CrossRef]
- Shi, H.; Lan, T.; Pinnavaia, T.J. Interfacial effects on the reinforcement properties of polymer-organoclay nanocomposites. J. Chem. Mater. 1996, 8, 1584–1587. [Google Scholar] [CrossRef]
- Rueda-Larraz, L.; d’Arlas, B.F.; Tercjak, A.; Ribes, A. Synthesis and microstructure–mechanical property relationships of segmented polyurethanes based on a PCL–PTHF–PCL block copolymer as soft segment. J. Eur. Polym. J. 2009, 45, 2096–2109. [Google Scholar] [CrossRef]
- Wen, X.W.; Pei, S.P.; Li, H.; Ai, F. Study on an antifouling and blood compatible poly (ethylene–vinyl acetate) material with fluorinated surface structure. J. Mater. Sci. 2010, 45, 2788–2797. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Banerjee, R. Biopolymer-based hydrogels for cartilage tissue engineering. J. Chem. Rev. 2011, 111, 4453–4474. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Tu, M.; Zeng, R.; Zhao, J. Preparation, characterization and cytocompatibility of polyurethane/cellulose based liquid crystal composite membranes. J. Carbohyd. Polym. 2012, 90, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- ASTM F 756-00. Standard Practice for Assessment of Hemolytic Properties of Materials; American Society for Testing and Materials: Philadelphia, PA, USA, 2000. [Google Scholar]
- Lönnroth, E.C.; Dahl, J.E. Cytotoxicity of liquids and powders of chemically different dental materials evaluated using dimethylthiazol diphenyltetrazolium and neutral red tests. J. Acta Odontol. Scand. 2003, 61, 52–56. [Google Scholar] [CrossRef]
- Barrioni, B.R.; de Carvalho, S.M.; Oréfice, R.L.; de Oliveira, A.A.R. Synthesis and characterization of biodegradable polyurethane films based on HDI with hydrolyzable crosslinked bonds and a homogeneous structure for biomedical applications. J. Mater. Sci. Eng. C 2015, 52, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Lönnroth, E.C. Toxicity of medical glove materials: A pilot study. Int. J. Occup. Saf. Ergon. 2005, 11, 131–139. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.A.R.; de Carvalho, S.M.; Leite, M.D.F.; Oréfice, R.L. Development of biodegradable polyurethane and bioactive glass nanoparticles scaffolds for bone tissue engineering applications. J. Biomed. Mater. Res. B Appl. Biomater. 2012, 100, 1387–1396. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Mn × 104(Da) | Mw × 104(Da) | PDI (Mw/Mn) |
|---|---|---|---|
| PCU-20% | 2.66 | 4.47 | 1.68 |
| PCU-25% | 1.37 | 2.08 | 1.52 |
| PCU-30% | 0.92 | 1.14 | 1.23 |
| PCU-35% | 0.88 | 1.06 | 1.20 |
| Samples | Tensile Modulus (MPa) | Ultimate Tensile Strength (MPa) | Elongation at Break (%) | Tensile Elongation (100%) Tensile Strength (MPa) | Tensile Elongation (300%) Tensile Strength (MPa) |
|---|---|---|---|---|---|
| 20%-PCU | 9.4 ± 1.4 | 35.6 ± 3.7 | 490 ± 50 | 8.7 ± 1.2 | 15.6 ± 2.1 |
| 25%-PCU | 12.6 ± 1.1 | 38.5 ± 1.2 | 510 ± 70 | 10.2 ± 0.8 | 18.4 ± 1.6 |
| 30%-PCU | 15.4 ± 3.1 | 30.5 ± 5.4 | 530 ± 30 | 10.3 ± 1.4 | 20.5 ± 1.2 |
| 35%-PCU | 18.3 ± 2.7 | 43.3 ± 2.3 | 380 ± 50 | 11.6 ± 1.7 | 24.6 ± 1.5 |
| Chronoflex® C | 14.8 ± 1.8 | 45.5 ± 3.1 | 410 ± 30 | - | - |
| Pellethane 2363-80A | 13.0 ± 2.0 | 34.0 ± 2.0 | 430 ± 20 | - | - |
| Samples | Average Optical Density | Negative | Positive | Hemolysis Ratio (%) |
|---|---|---|---|---|
| 20%-PCU | 0.7175 | 0.0437 | 0.3642 | 2.1024 |
| 25%-PCU | 0.6990 | 0.0437 | 0.3642 | 2.0446 |
| 30%-PCU | 0.5339 | 0.0437 | 0.3642 | 1.5296 |
| 35%-PCU | 0.5104 | 0.0437 | 0.3642 | 1.4560 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, R.; Wang, X.; Yang, J.; Wang, Y.; Zhang, Z.; Hou, Y.; Lin, F.; Li, Y. Influence of Hard Segments on the Thermal, Phase-Separated Morphology, Mechanical, and Biological Properties of Polycarbonate Urethanes. Appl. Sci. 2017, 7, 306. https://doi.org/10.3390/app7030306
Zhu R, Wang X, Yang J, Wang Y, Zhang Z, Hou Y, Lin F, Li Y. Influence of Hard Segments on the Thermal, Phase-Separated Morphology, Mechanical, and Biological Properties of Polycarbonate Urethanes. Applied Sciences. 2017; 7(3):306. https://doi.org/10.3390/app7030306
Chicago/Turabian StyleZhu, Rong, Xinyu Wang, Jing Yang, Yiyu Wang, Zongrui Zhang, Yuanjing Hou, Fei Lin, and Yi Li. 2017. "Influence of Hard Segments on the Thermal, Phase-Separated Morphology, Mechanical, and Biological Properties of Polycarbonate Urethanes" Applied Sciences 7, no. 3: 306. https://doi.org/10.3390/app7030306
APA StyleZhu, R., Wang, X., Yang, J., Wang, Y., Zhang, Z., Hou, Y., Lin, F., & Li, Y. (2017). Influence of Hard Segments on the Thermal, Phase-Separated Morphology, Mechanical, and Biological Properties of Polycarbonate Urethanes. Applied Sciences, 7(3), 306. https://doi.org/10.3390/app7030306